Genetica 95: 173-193, 1995
THE EPIDEMIOLOGY AND TRANSMISSION OF AIDS: A HYPOTHESIS LINKING BEHAVIOURAL AND BIOLOGICAL DETERMINANTS TO TIME, PERSON AND PLACE
Gordon T. Stewart
Emeritus Professor of Public Health, University of Glasgow, Glasgow G12 8QQ, UK
Present address: Glenavon, Clifton Down, Bristol BS8 3HT, UK
Abstract
Epidemiologically, the Acquired Immune Deficiency Syndrome, AIDS, is transmitted and distributed in the USA and Europe almost entirely in welldefined subsets of populations engaging in, or subjected to, the effects of behaviours which carry high risks of genital and systemic infections. The persons predominantly affected are those engaging in promiscuous homosexual and bisexual activity, regular use of addictive drugs, and their sexual and recreational partners. In such persons and in subsets of populations with corresponding lifestyles, the risk of AIDS increases by orders of magnitude. Because of continuity of risk behaviour and of associated indicator infections; the incidence of AIDS over 35 year periods is predictable to within 10% of actual totals of registered cases in the USA and UK. Secondary transmission of AIDS beyond these groups is minimal or, in many locations, absent. There is no indication of appreciable spread by heterosexual transmission to the general population.
The Human Immunodeficiency Virus, HIV, is transmissible to some extent in general populations, and more so among promiscuous persons. It may cause viraemia, lymphadenopathy and latent infection (HIV disease) in anyone. In persons engaging in risk behaviours which themselves alter or suppress immune responses, it can interact with MHC, antibodies to other organisms and to semen, and other allogenic antigens to initiate a programmed death of CD4 lymphocytes and other defensive cells, as in grafthost rejections. This occurs also in haemophiliacs receiving transfusions of blood products, and is more pronounced in persons with reactive HLA haplotypes. The susceptibility of particular subsets of populations to AIDS is thereby largely explained. But these changes occur in the absence of HIV, and so do Kaposi's sarcoma, lymphadenopathies and opportunistic infections which are regarded as main indicators of AIDS. The hypothesis that HIVI can do all this by itself and thereby cause AIDS is falsifiable on biological as well as epidemiological grounds.
An alternative hypothesis is proposed, linking the incidence of AIDS to the evolution of contemporary risk behaviour in particular communities and locations in the USA, UK and probably in most of Europe. It does not pretend to explain the reported incidence of AIDS in Africa and other developing regions where data are insufficient to provide validation of the pattern of disease and contributory variables.
The immediate, practical implication of this alternative hypothesis is that existing programmer for the control of AIDS are wrongly orientated, extremely wasteful of effort and expenditure, and in some respects harmful.
Introduction
In 1984, a US Secretary of State announced that a retrovirus then named HTLV3, isolated from one patient in Paris, was the unique cause of loss of immunity in a severe infectious disorder first described in 1981 in some homosexual men and drug addicts admitted with unusual symptoms to hospital in New York City and California. Since then, there has been a world wide consensus in medical science and beyond that this retrovirus, renamed HIVI, and its innumerable variants are the unique cause of a more complex and extending range of disorders classified and repeatedly reclassified as the Acquired Immune Deficiency Syndrome, AIDS. When this consensus was challenged by P.H. Duesberg in 1987 (see below), R.C. Gallo, the chief proponent of the claim for HTLV3, and leaders of the consensus replied in a Forum reported by Science Magazine (1988;241;514) that the 'Strongest evidence that HIV causes AIDS' came less from isolates of the virus itself than from prospective epidemiology. Having been engaged in epidemiological work which suggests the reverse, I then began a long struggle to persuade the immense epidemiological sector of the consensus that this assertion, no less than the microbiological evidence of causation of AIDS by HIV, merited reconsideration.
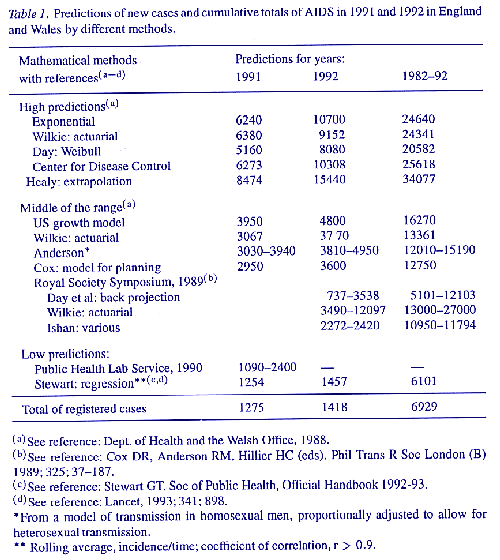
When Duesberg challenged the HIV hypothesis in 1987, the epidemiological evidence depended essentially upon a correlation between antibodies to HIV and a diagnosis of AIDS. This was a circular argument since, after 1984, seropositivity to HIV mandated a diagnosis of AIDS in patients with a scheduled range of diseases, whether or not they were in risk groups. In 1989, a formidable defence of the epidemiological consensus was presented by the Royal Society of London in a Symposium (Cox, Anderson & Hillier, 1989) of invited papers which analysed the epidemic of AIDS to date in the UK and made predictions about its incidence through 1992. These analyses and predictions, like those of a preceding official Report (UK Dept. of Health and the Welsh Office, 1988), rested upon assumptions not only that HIV was the essential cause of AIDS in all its forms but also that all who were seropositive would get AIDS which would then cause epidemics of tens of thousands of cases by heterosexual transmission in the general population of the UK.
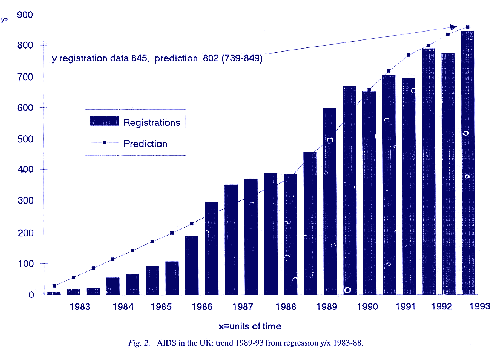
The assumption that HIV would spread by heterosexual transmission was, to a limited extent, correct but predictions of similar spread of AIDS, as in the London Declaration of 1988, from the WHO (Mann, 1989; Sato, Chin & Mann, 1989) and by the Advisory Committee on Dangerous Pathogens (1990) were numerically exaggerated (Table 1), as were actuarial and other projections used for planning and estimates of spread outside the original risk groups of homosexual men and drug addicts. They were particularly wrong (Table 2) in the prediction that heterosexual transmission of AIDS in the general population would give rise to a general epidemic (Anderson & May, 1987; Public Health Laboratory Service, 1990). What the epidemiological evidence did show in the USA, in the UK, and most of Europe and Australasia, was a continuing increase of AIDS in the original high risk groups. This trend is so consistent that, even in New York City, an original epicentre of AIDS, a regression model (Stewart, 1992a, 1993a) of incidence over units of time since 1983 predicted in 1989 a cumulative total of 45,487 cases by the end of 1992 (N = 44,231). Predictions for the intervening years were correct to within 10% of registered cases though, in 1993, there was a departure from linearity caused entirely by reclassification of AIDS by the CDC in that year (see below) to include all cases of carcinoma of the cervix, tuberculosis and many bacterial pneumonias in HIVseropositive patients (Fig. 1). In the UK, with a much lower incidence, regression models through 1993 and to date are equally accurate
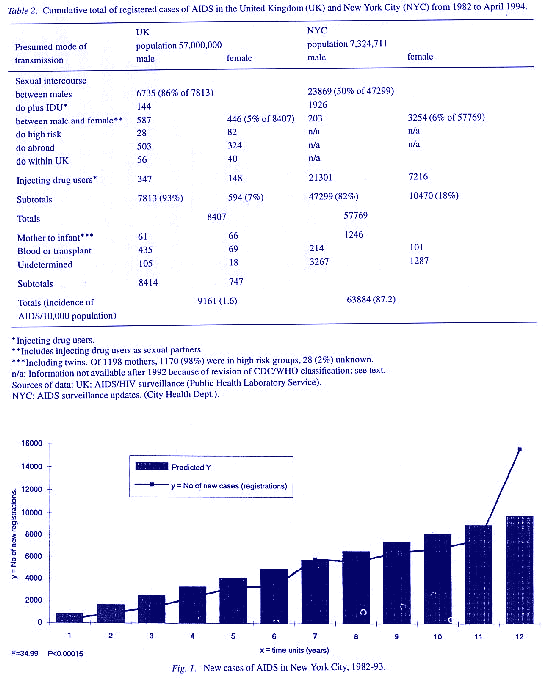
(Fig. 2). AIDS is a disease which began and spread in males. In females and their infants outside risk groups, the incidence of AIDS after ten years is negligible in the UK as in New York City (Table 2).
All of this is verifiable in the registrations published by official surveillance in the USA and UK. The epidemiological evidence in New York City does, however, indicate an increase of AIDS by presumed heterosexual transmission in men and women in the blackHispanic ethnic minorities in parts of Manhattan, Brooklyn and Queens, especially if they use drugs (Stoneburner et a/., 1990). To investigate the role of HIV in this, Pagano et al. (1991) used an elaborate model with three levels of seroprevalence to estimate the incidence of AIDS through 1995. Their estimates for 1991 ranged from 16,106 assuming no new infections to 29,962 assuming 10,000 new infections annually (N = 6,800). Fordyce et al. (1991) estimated the cumulative incidence of AIDS in female partners of drug addicts to be 3,900 (N = 4091) from formulae based on conditional probabilities of uninfected women acquiring HIV from infected male partners. But if P/ivdu, the probability that a woman in New York will have a drug user as a sex partner, is replaced in the model by the alternative probability P/hiv that she is equally or more likely to be infected by any man who is HIVpositive, the estimate of AIDS again becomes exaggerated.
Incidence in the UK expressed as a rolling average or periodically since 1982 (Stewart, op. cit.) and despite considerable fluctuations in reporting follows the same close correlation with units of time (r = 0.97). Regression accurately predicted about 3,000 cases by December 1989 (N = 2,830), 1,254 in 1991 (N = 1,275), 1,365 in 1992 (N = 1,418) and in cumulative total, excluding visitors, of 6,540 (N = 6,929, including visitors) by the end of 1992 (Tables I and 2, Fig. 2). This is very much less than the range of 3,810 4,950 predicted by Anderson (2) for the year 1992 which gave a cumulative total, for planning, of 12,010 15,190 cases in the UK by December 1992 and much higher totals thereafter. In contrast, the regression model predicted only 1,554 new cases (excluding visitors) in 1993 when the actual total (including visitors) was 1,619, to give a cumulative total by 1994 of 8483 cases (N = 8529), over 3,000 less than the lowest official estimate for 1992.
The large errors in these predictions In Britain and in the USA are inherent. They derive from acceptance of the hypothesis that continuing sexual transmission of HIV is the main determinant of the incidence of AIDS. This led to further assumptions that heterosexual transmission will increase until females are equally at risk, that all who are infected with HIV will develop AIDS sooner or later and die, and that mathematical models based on this reasoning will predict the course of events correctly, within wide limits. The passage of time and the data since 1989 in the USA and in Britain falsify all this (Tables I and 2).
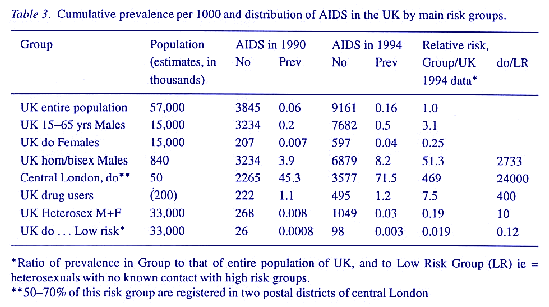
So also do backprojections for the UK, projections by Bregman and Langmuir (1990), based on Farr's Law of Epidemics, for the USA and estimates using sociogeographic data in New York City (Wallace, 1991). But predictions based on regression equations (Figs. I and 2) are more reliable because they leave less than 10% of residual variance in incidence since 1983 unexplained. Such accuracy in a deterministic model is highly unusual. The most obvious reason is that transmission and expression of AIDS is restricted to subsets of the population which engage in, or are exposed to, the effects of continuing riskbehaviour (Stewart, 1990a, 1991, 1992a). Even in those subsets, the incidence may now be decreasing in the USA and the UK. The accuracy of predictions from raw data by regression methods is consistent with the finding that, while HIV1 has already spread widely in both sexes because of its presence in genital secretions and in needles shared by drug addicts, the occurrence of AIDS beyond risk groups, as predicted in al1 of ficial surveys, is low or absent even in communities where HIV 1 is prevalent. The differences in levels of risk, calculated from best estimates of relevant denominators, are enormous (Table 3) and need much more attention.
The obvious conclusion is that, in such communities, AIDS cannot develop unless other interacting causes of immunosuppression and cachexia are present. Registration data from the USA since 1985 show that prevalence of HIV and AIDS is strongly influenced by ethnic, urban, economic, social, occupational and mobility factors. In New York City, the situation is dominated by the fact that 60% of adults and 90% of infants with AIDS are in Black or Hispanic communities and by the 33fold difference in rates of disease, from < 50 to > 1,500 per 10,000 adults, in different districts of the city (Table 2). In the UK, analytical data are much less precise but, to 30th June 1992, 1,354 cases of AIDS (38% of the cumulative total for Britain) were reported from NW London; 94% of these cases were male and 91 % of them homosexual. Despite this prevalence of AIDS and also of HIV seropositivity, at least 20 times higher than the remainder of Britain, there were only 31 cases of AIDS in women transmit ted by (presumed) heterosexual intercourse out of 401 who were seropositive. Infants born in London in 1992 were much more often seropositive than those born outside but, because surveillance is voluntary, unlinked, anonymous and confidential, it is impossible to know how this relates to the risks, status and treatment of the 50 mothers who have given birth from 198293 to babies who developed AIDS (about 1:120,000 births). In southeast England generally, it was estimated that 92% of neonatal seropositives and 7 out of 8 babies who developed AIDS in the first year of life came from mothers who were born in Africa (Aces, Parker & Cubitt, 1992). There is clearly a pressing need for a more informative data base and correlation matrix in the UK but, even in these limited data, it would appear that there is no appreciable spread of AIDS to infants from their mothers in the general population outside specified risk groups in or from welldefined locations.
The current hypothesis
The hypothesis that HIV is the unique cause of AID is an inductive generalisation based on a few agree facts and an acceptance in medical, sociological an political circles of corroborative reasoning, conjectur and consensus. The facts (BarreSinoussi, Cherman & Rey, 1983; Gallo, Salahuddin & Popvic, 1984; Dal gleish et al., 1984; Levy & Chimabukuro 1985; He Pomerantz & Kaplan, 1987; Hanafusa, Pinter & Pull man, 1987; Gallo, 1987) are that (i) HIVs can be isolated from, or identified by biochemical probes in celh blood and secretion of an (unknown) proportion c patients with AIDS; (ii) in patients with AIDS wh are tested serologically, antibodies specific for antigens prepared from envelopes of the original isolate of LAV 1/HTLV III are usually detectable; (iii) in term of this test, there is a correlation between the presenc of HIV and AIDS in a community; (iv) HIVs appec to be transmitted from persontoperson by anal an vaginal intercourse, or parenterally via infected needles or blood transfusion, or congenitally; and (v) HIVs have high affinity for, and fuse with specific CD4 membrane receptors on helper Tlymphocyte and other mononuclear cells, transcribe their RNA int the DNA of the cells' nuclei and form virions which can infect other Tlymphocytes. The reasoning (Ho Pomerantz & Kaplan, 1987; Blattner, Gallo & Tenil 1988; Institute of Medicine, 1988; Baltimore & Feir berg, 1989; Fauci, 1988) is that HIVs can thereby weaken or destroy cellmediated immunity, and that persons thus affected always or almost always succumb to a specific syndrome of generalised immune deficiency which then renders them susceptible to other, opportunistic infections and to various disorders of lymphoid cells and vital processes with fatal or nearfatal results. The conjecture of these authors and very many others is that infection with HIV is necessary and sufficient to explain this pathogenesis, irrespective of riskbehaviour. The consensus of the medical and scientific establishment, and practically all health authorities is that epidemiological evidence and predictions support this reasoning, and that any departure from it is heresy, a threat to public safety and efforts to control a dangerous epidemic, and to dedicated research.
Weakness in the HIV hypothesis
Despite this overwhelming consensus, or perhaps because of it, there are many uncertainties and flaws of reasoning in this hypothesis on epidemiological, clinical and microbiological grounds. Epidemiologically, the data presented above falsify the assumption that AIDS is spreading in general populations in the USA and UK by heterosexual transmission of HIV. The salient point that AIDS was described (Gottlieb, 1981; and see report from the CDC, Atlanta, Gal, from California State Health Department and from New York City, Dept. of Health) simultaneously in California and in New York City as focal incidents, with no evidence of anything comparable elsewhere at that time, is often overlooked. The first cases were registered in San Francisco on July 1st, 1981. By March 1985. 1.000 cases had been registered, of which 992 (99%) were male and 98% homosexual or bisexual with multiple partners, with a very high prevalence of gonorrhoea, syphilis, hepatitis and other sexuallytransmissible infections, with 13% using intravenous drugs and 98% resident in the Bay area. The position in 1992, in the UK, most of Europe, Australasia and North America at least, is that AIDS is still predominantly a disease of men (Table 2) and that the women who acquire it, at a much lower incidence, are those who expose themselves to high risks of infections from partners with AIDS or at risk of AIDS because of homosexual and bisexual behaviour, and from use of toxic drugs.
In female prostitutes, who are a risk group for any sexuallytransmitted disease (STD), HIV infection and AIDS are prevalent in African cities (see WHO: weekly epidemiological reports) but not in North America or Europe unless they use drugs habitually or have other STDs. Seale (1988) suggested that, for this reason, AIDS did not qualify clinically for classification as an STD. and also because it is essentially 'A bloodborne infection which is transmitted only with considerable difficulty during biological sexual intercourse' from which he excluded penileanal intercourse. This would be consistent with the findings, in multicenter prospective and many other studies (Marmor, FriedmanKien & Laubenstein, 1982; Shilts, 1987; Gunzburg, Fleming & Millar, 1988; Detels, English & Visscher, 1989; Ma & Armstrong, 1989; Beral, Bull & Darby, 1990) of homosexual men, that rectal trauma and infections from bleeding, douching, fisting and other traumatic and anoerotic acts are associated with progression to AIDS and ARCs.
In Africa, the Caribbean and Asia, notifications of seropositivity to HIV and of AIDS to the WHO are increasing sharply. The epidemiological and clinical patterns, at face value, are different from those of the western world. Cases are reported with equal frequency in males and females and homosexuality and use of drugs are uncommon as risk factors. In place of the opportunistic infections reported in developed countries, tuberculosis, diarrhoeal diseases, malnutrition, exhaustion and early death are the main clinical features. Occurring as they do on a considerable scale in young men and women, this is widely regarded as a new, uncontrollable epidemic in many subSaharan countries. Details of CD4 counts, isolation of HIV and other tests are seldom available.
There is considerable genetic divergence in the comparatively small number of strains of HIV isolated in third world countries (Louwagie, McCutchan & Van der Groen, 1992; Pfutzner, Dietrich & von Eichel, 1992). As in the USA and Europe, AIDS is very uneven in distribution. Originally, it was reported from Uganda (Serwadda, Sewankambo & Carswell, 1985) as a localised, wasting 'Slim' disease, but now it has become an acute infection strongly linked with tuberculosis (KonoteyUhulu, 1989; Berkley, WidiWirski & Odware, 1989; de Cock et al., 1992). Along with other STDs, AIDS is increasingly prevalent in certain cities on the international travel routes of persons who sample the local risks, and convey their own infections and riskbehaviour to local populations. Hence the ominous spread in Africa via truckroutes, and in the UK in certain immigrants, visitors, returning travellers and their domestic partners (Hawkes et al., 1992).
AIDS was not described in Africa until 1984, some years after the first occurrences in white men in the USA and in black Haitian immigrants in New York City. This, together with the prevalence of seropositivity to HIV in unconfirmed ELISA tests in Zaire in 1985, led the consensus to the belief (Gallo, 1987; Mann, 1989) that AIDS had therefore originated in Africa. A wide search was therefore made to find support for a subsidiary hypothesis that AIDS had spread somehow from Africa to the USA, if not to the rest of the world. The reasoning is that HIVs evolved like Simian Immune Deficiency Viruses (SIVs) latterly identified in nonhuman primates in Africa, either by phylogenetic separation of a retrovirus from a common progenitor in the distant past, or by crossspecies transfer to humans more recently (McClure & Schultz, 1989). If this is shown to be genetically plausible, or if immune deficiency occurs naturally in nonhuman primates infected with SIVs, or artifically with HIV in the absence of other infections, the argument might become credible. But a very extensive search has revealed no common progenitor, and no link between any SIV and HIV 1 in Africa (Scale, 1988). Irrespective of the simian or other origins of HIVs the view that AIDS in the clinical pattern now observed was already prevalent in Africa is entirely speculative. There is no comparable study of the alternative possibility that AIDS might have travelled from its observed origin in the USA in 1981 somehow to Africa.
Classification of AIDS
AIDS is registered internationally as if it were a single infectious disease, and is surveyed accordingly, that is to say as if it were a selfdefining dependent variable. But the original empirical classification by the US/CDC accepted internationally in 1983 was expanded in 1987 to schedule a range of neoplasms, infections, malnutrition and dementia in which seropositivity to HIV, with or without riskgroup identification, or these symptoms without seropositivity in risk groups were made eligible for classification as AIDS. This increased the size of the epidemic in the USA by about 27%. To this list of 'indicator' diseases, a further revision in 1992, synchronous with identical changes in the International Classification of Diseases, added cancer of the cervix, tuberculosis and other diseases in persons who are seropositive. This has already added numerous females to the incidence of AIDS (Fig. 1) and manufactured an epidemic in women which is, otherwise, conspicuous by its absence (Stewart, 1992b). To add to the confusion, there are growing doubts (Couruce, Muller & Richard, 1986; Meyer & Panker, 1987; Midthum, Garrison & Clements, 1990; Mortimer, 1991; Davey, Dayton & Metcalf, 1992; PapadopulosEleopoulos, Turner & Papadimitriou, 1992) about the lack of an independent goldstandard validator, and therefore about the specificity of serotests for HIV, even with double testing by immuneabsorption and blot, in the presence of other infections and immunological disorders, especially in tropical countries. The fall in CD4 lymphocytes, required for validation, but seldom performed outside specialised units, is also a nonspecific event (Drew, Mills & Levy, 1985; Jason, Holman & Evatt, 1990; RootBernstein, 1993) which occurs in many other infections and after infusions of foreign protein, e.g. to haemophiliacs (Carr, Edmond & Prescott, 1984).
Clinical diagnosis
In centres with expertise and facilities, surveillance of AIDS is monitored, with appropriate checks and tests. In reports from such centres, the pattern of AIDS is consistent and predictable, as above. But HIVs can be isolated from persons with and without AIDS or related conditions (ARCs). The great majority of seropositives show no signs of disease. To accommodate the continuing absence of AIDS in such persons, the 'incubation' period between infection and the onset of disease has been extended to 15 years or more in the 1987 and subsequent classifications by the CDC and WHO. This had led to a muddled situation in which anyone with the wide spectrum of symptoms and signs in these various classifications who has antibodies to antigens in any one of the several antigenkits, or a fall in CD4 counts, is liable to be diagnosed as AIDS: so are person in risk groups with some of the signs, whether or not they are seropositive, and seronegatives not in riskgroups with signs who then become seropositive. The Revised Classification now in use raises to 28 (in Section B2024) the number of independent diagnoses that may now be registered as AIDS. Section B21.09 includes 6 specified and 3 unspecified malignancies, to which cancer of the cervix is now added. This alone has already added thousands of cases of AIDS to survey totals of AIDS internationally. Section B21.79 (any cancer) and B22 (any other specified or wasting disease) in anyone who happens to be seropositive and in many who are not, add many more. Details of collateral or coincidental disease, and tests to exclude other diagnoses are not required for registration, not even the CD4 (T4) lymphocyte count, mandated by the consensus as the hallmark of AIDS. Against all logic, this extraordinary diagnostic gallimaufry is accepted by the medical profession and the consensus as input data, not only for surveillance, but also for prediction of the spread of AIDS internationally.
Microbiology
HIVs have several properties which may relate to the pathogenesis of AIDS (Gallo, 1987, Fauci, 1988; Evans, 1989a). In addition to thegag, pol and env genes common to all retroviruses, they have five or more nonstructural genes including a tat masterswitch (Carlin, Peterlin & Derse, 1992; Elangovan, Subramanian & Chinnadurai, 1992) which enable them to regulate their replication and to delete, insert and duplicate nucleotides so as to evade immune responses. They are naturally lymphotropic and enter Thelper lymphocytes, among other cells, because their envelope glycoproteins interact with specific CD4 surface receptors (Dalgleish, Beverley & Clapham, 1985). In situ, they transcribe their RNA into the cell's DNA and replicate to form virions which have been shown to infect monocytes and macrophages as direct transfer (Fauci, 1988; Li & Burrell, 1992; Innocenti, Ottoman & Morand, 1992). These migrate to other sites, including the brain and thymus, where HIV may infect other cells, remain latent or replicate in accordance with the interplay of positive and negative regulatory elements in its own genome, or in those of defensive cells.
Retroviruses are characteristically latent (Duesberg, 1987; and see Hanafusa, Pinter& Pullman, 1989). HIV is no exception in the great majority of infected persons who remain, to date, asymptomatic. But this unusual combination of genetic heterogeneity and antigenic variability between strains of HIV, with mutability of nucleotide sequences within strains and tropism for migrant cells, would seem to offer plausible mechanisms for activation. In a search for amino acid sequences involved in cell tropism, Cheesebro et al. (1992) found homology in macrophagetropic clones from different patients. Tcelltropic clones were, in contrast, highly heterogeneous. Sitespecific mutations in amino acid sequences in the V3 region of HIV isolates appeared to be responsible for these tropisms. This hypervariable domain within gp 120 is recognised (Shioda & Levy, 1992) as a major determinant of the ability of HIVI to infect Tcells and macrophages. These and other examples of genetic heterogeneity occur within as well as between strains, so much so that HIV has been described as a quasispecies in which no two genomes are identical (WainHobson, 1989; Vartamian, Meyerhans & WainHobson, 1992). Many are defective and, by the same token, noninfective. Proviral sequences vary accordingly, in the same or in successive isolates, and so do replication rates in vivo, as disease advances. The consensus accepts these properties of HIV as explanations of its ability to emerge from latency and of its pathogenicity. The impetus of over 60,000 supportive papers since 1983 is formidable even though much of it depends upon results obtained and elaborated in vitro. There is, as in other branches of biomedical research, a greater focus on the behaviour of the microbe than on that of the host.
The only prominent retrovirologist to question the consensus about HIV is P.H. Duesberg of the Department of Molecular Biology at Berkeley, CA, whose dissent is absolute. He considers that HIV is as inactive in patients with AIDS as it is in asymptomatic carriers. In fact, he goes further and rejects the wider claim by most virologists that latent viruses and mutated genes can be pathogenic (Duesberg & Schwartz, 1992). His arguments, dating from 1973 and extending far beyond the virology of AIDS, are set out in papers (1987, 1989a) and responses to criticisms (1989b, c). His main argument, as a retrovirologist, is directed against the central dogma that HIV infects, multiplies in and kills enough Thelper lymphocytes to destroy immunity. His observations that, in patients with AIDS, only 1 in 500 Tcells ever contain a provirus of HIV and that HIV cannot kill these cells have been confirmed independently by Lemaitre et al. (1990), the team which originally isolated HIV. His assertions that neutralising antibodies restrict multiplication of HIV, and that it does not have the biological energy or biochemical capacity to produce pathological changes in vivo have been disputed (Blattner, Gallo & Tenin, 1988; Baltimore & Feinberg, 1989; Evans, 1989a, b; Weiss & Jaffe, 1990) but not falsified. He offers evidence that, when latent viruses are reactivated after neutralisation by antibodies, this is due to independent factors (other infections, immunosuppressive conditions) and not to mutations in the coding region of the virus. He applies this view to other retroviruses and diseases no less than to HIV. If he is correct, or even partially correct, the implications will be revolutionary, for they will dismiss as circumstantial the current beliefs that latent viruses cause specific, progressive infections and that mutated oncogenes can ever cause cancer. HIV would then be merely a marker for AIDS while cancers would be more likely to arise from clonal chromosomal abnormalities.
In his original (invited) contribution to this field in 1987, Duesberg made a critical analysis of the facts and gaps in retrovirology in this regard. But in rejecting HIV as the cause, he also attacked the core of the biomedical research on AIDS. This led, after a considerable delay, to polarised reprisals with a minimum of reasoned debate (Blattner, Gallo & Tenin, 1988; AAAS Policy Forum, 1988) on his main question about the cytopathic effect of HIV. This is still unresolved. Even so, Duesberg would be wrong in rejecting a pathogenic role for HIV on this account alone because as Cheesboro et ai. (1992), among others, have shown it can unquestionably infect monocytes and macrophages by celltocell transmission, without killing them, and then travel in them to other tissues where the presence of infected cells would be enough to arouse inflammatory response and hence disease, e.g., a glial respcnse in the brain. If there was already a latent infection with Toxoplasma, this could multiply to cause encephalopathy which is reported in about 30% of patients with cerebral AIDS (RootBernstein, 1993). When definitive signs of AIDS develop, HIV replicates and releases antigen (Ho, Pomerantz & Kaplan, 1987). It is insisted (Fauci, 1988; Baltimore & Feinberg, 1989; Weiss & Jaffe, 1990) that this is due to a regulated change from latency to accelerated replication, but it might equally be part of the general multiplication of organisms which occur in any immunosuppressive state, and is a main contributor to death in AIDS.
Duesberg's rejection of the claim that HIV kills lymphocytes per se is supported by the work of Lemaitre, Montagnier and their colleagues (1990) showing that the cytocidal effect of two archetypal strains of HIVs (LAVBru of HIVI and Rod of HIV2) in vitro was lost in the presence of noninhibitory concentrations of tetracycline analogues. Since these compounds do not interfere with the infectivity of HIVs, the likelihood is that a tetracyclinesensitive organism, now confirmed as a mycoplasma, plays the role of synergistic cofactor in HIVinduced cell Iysis. It is now thought to be identical to Mycoplasma fermentans (incognitas) previously isolated by Lo (1986) from patients with AIDS. This mycoplasma has been visualised, isolated in culture and identified by DNA probes in thymus, lymph nodes, spleen, liver, brain and placenta of patients with AIDS and from HIVnegative patients with fulminant necrotising lesions or fatal disease in these organs. Most of the isolates were made from tissues without necrotic or inflammatory changes but ultrastructural examination showed, in some cases, intracellular mycoplasma and cytopathic changes in lesions from which no other pathogens were visualised or isolated. When injected into four monkeys, the mycoplasma caused systemic infection followed by wasting and death in 79 months with necrotic lesions without inflammatory reaction in which the mycoplasma (originally thought to be a viruslike agent VLIA) was identified by immunochemistry, in situ hybridisation and electron microscopy (Lo, Wang & Newton, 1989). DNA from M. fermentans has been detected in the blood of seropositive patients (Hawkins et al., 1992) but, in a systematic study of patients attending an STD clinic, Katseni, Gilroy and Ryait (1993) found it also in peripheral blood mononuclears, throat swabs and urine from a majority of seronegative as well as seropositive homosexual men. This is unrelated to the stage of disease, CD4 count and cellular HIV load in the seropositive subjects.
Mycoplasmas are notorious as contaminants in tissue cultures, especially those requiring reinforcement. It is surprising that they have not been reported in the innumerable other laboratories which are working round the clock internationally with HIVs from many sources. Coming as they do from the scientists who discovered HIV, the results quoted above prove that HIV cannot kill Tlymphocytes without assistance from a mycoplasma. The work of Lo and his colleagues suggests, but does not prove, that this organism may have a synergistic or pathogenic role in AIDS.
The image of HIV as a universal pathogen is weakened further by the fact that the strains isolated in the USA and Britain were merely subcultures of the original strain (LAVI) isolated in 1983 from a gland from a patient in Paris. Genuine independent isolates show continuous diversification (Shioda & Levy, 1992; Vartamian, Meyerhans & WainHobson, 1992; Spencer, et al., 1994). The strain type HIV1 is uncommon or absent in some populations with AIDS in Europe and Africa, and is not crossreactive with some prevailing strains (Quinn, Piot & MacCormick, 1987; Zwart, de Jong & Wolfs, 1990). Genetic heterogeneity and divergent subtypes of HIV on a wider scale, as reported above from Africa and India, might explain the variability in symptomatology and progression of AIDS. By the same token, this means that some strains are likely to be less, or much less pathogenic: a prospect already verified by the survival of the majority of infected persons in the USA and Europe. The consensus views that all who are infected will develop AIDS and probably die is falsified on this score alone.
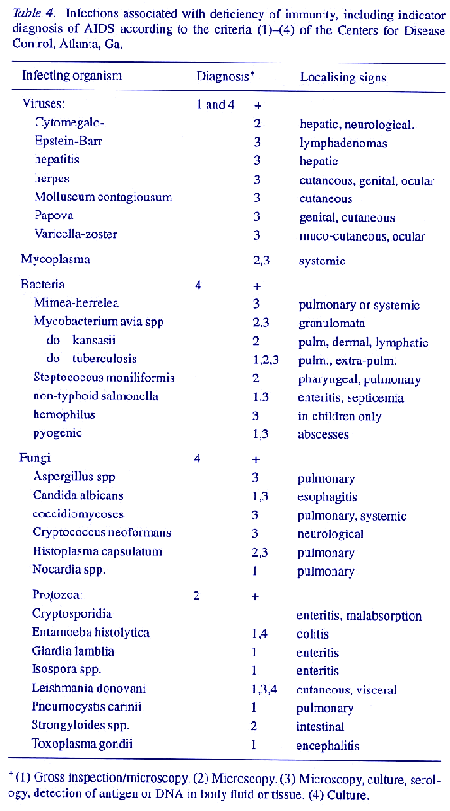
In the weeks after infection with HIV, some persons develop a brief illness with fever and lymphadenopathy, similar to infectious mononucleosis. Seroconversion then occurs, producing antibodies to HIV envelope proteins gpl20 and p24. The majority of infected persons remain asymptomatic but a minority, identifiable in terms of riskbehaviour and exposure to further infections, show the hallmark fall in CD4 lymphocytes with reversal of the T4/T8 ratio, lymphadenopathy, energy, loss of immunity, multiplication of infection organisms including HIV, and other signs of ARCs or AIDS. From then on, AIDS seems to be irreversible, despite specific antiviral and symptomatic treatment. The main pathological findings are pneumonias (Pneumocystis carinii or acute bacterial), gastrointestinal infections (Candida, salmonellae, shigellae, entamoebae), coincident or secondary infections of the skin, viscera and brain with other pathogenic and opportunistic bacteria, fungi and protozoa (Table 4); and, mainly in homosexual men, Kaposi's sarcoma. Some or all of these, along with failing nutrition and exhaustion, lead usually to early death, though mortality since 1983 has decreased considerably in developed countries (D'Arminio, Vago & Lazzarin, 1992).
Haemophilia
The occurrence of serpositivity to HIV and of AIDS following transfusions of infected blood and blood products in some haemophiliacs and other patients, and the apparent cessation of this after donors were screened and blood sterilised by heat, have been advanced as selfevident and conclusive proof of the causation of AIDS by HIV (Tsoulkas et al., 1984; Lud1am, Tucker & Steel, 1985; Hiltgartner, 1987; Ward, Bush & Perkins, 1989; Darby, Rizza & Doll, 1989).
The existence of seroconversion and signs compatible with AIDS in recipients of transfusions, especially in haemophiliacs, is not in doubt. But this has to be considered against background facts. Blood transfusions are, by themselves, wellknown to be temporarily immunosuppressive. Patients receiving frequent transfusions are, by definition, in a risk category. Mortality, even in the short term, is often high, inde
Homosexual and bisexual behaviour
Male homosexual relationships without anal intercourse or injections of drugs arguably the majority (Ma & Armstrong, 1989; Stewart, l990b) are not associated with AIDS; neither is lesbianism. AIDS began and prevailed among those who are still at highest risk, namely the passive male, and sometimes female, recipients of anal intercourse. This is because the rectal mucosa and its supporting tissues are relatively fragile, designed for excretory, not intrusive activity. When the thin submucosa is eroded and blood vessels damaged, the tissues and blood stream are opened to invasion by all the organisms of the faecal microflora, by the pathogens of all the sexually transmitted diseases, and many others. The risk of trauma and infections increases greatly with the frequency, variety (oroanal, linguavaginal) and violence of the sexual activity and preference, as with 'fisting' and other accessory, traumatic and contaminating procedures, and with multiplicity of partners (Wilkins & Sonnabend, 1983; Mavligit, Talpag & Hsia, 1984; Moss, Osmond & Bacchetti, 1987; Winkelstein, Wiley & Padian, 1988; Sonnabend, 1989).
In such persons, the unregulated use of antimicrobial drugs for selftreatment of gonorrhoea and other infections inhibits the competitive flora of the intestine, opening it to bacterial and fungal superinfections which are indicator conditions for diagnosis of AIDS. Notable among these are Pneumocystis carinii, Candida albicans, cryptosporidia and organisms causing chronic diarrhoca, and hence dehydration, loss of electrolytes and exhaustion. The frequent presence of semen in the rectum and blood adds allogenic, 'nonself' reactions which dysregulate immune responses (Witkin & Sonnabend, 1983; RootBernstein, 1993). The faecal microflora interacts with semen to form Nnitroso compounds, some of which are immunosuppressive and carcinogenic (Schoental, 1988). Immunosuppression also occurs (Newell, Mansell & Spitz, 1985; Mirvish & Haverkos, 1987; Vandenbrone & Pardoel,1989) from the use of volatile alkyl nitrites (poppers) as aphrodisiacs and relaxants an effect which conveniently extends to the rectal sphincter. In experimental animals, these nitrosating agents are lymphotoxic, causing immunosuppression followed by death from acute and chronic infections. Surviving animals sometimes develop lymphomas (Schoental, 1988). It is possible that the conjunction of Nnitroso compounds from semen with volatile nitrites contribute to Kaposi's sarcoma which occurs in this context independently of
HIV (Beral, Bull & Darby, 1990). All this, combined with frequent, promiscuous anal and bisexual intercourse with dozens or hundreds of partners had become a way of life in the dedicated communities in which AIDS was first observed and in those (of remarkably similar persuasions and microflora) to which it quickly spread, internationally. Knowledge of the dangers sometimes led to a reduction in riskbehaviour but, by this time, genital and other infectious diseases were accepted as features of their way of life.
Transmission in heterosexuals
This is regarded by the WHO and the consensus as the usual mode of transmission of AIDS in many third world countries. In North America and Europe, surveillance shows some increase in AIDS occurring in both sexes from presumed heterosexual transmission but, in the UK and USA at least, the increase is fractional, even in persons with high risk partners (Tables 2 and 3) and in those attending STD clinics, and is confined to major urban areas. It is uncommon in prostitutes unless they are using drugs. AIDS in women outside the main risk groups is minimal or zero. Since seropositivity to HIV in random samples may be equal in the sexes, and since sexual intercourse with more than one partner by the age of 18 is now common, the key questions arising from the absence or infrequency of AIDS in females are if and why it occurs at all. Confidentiality of records and lack of contacttracing deemed necessary in other STDs preclude answers. How much of what there is has been acquired from bisexual men, from anal intercourse, from undeclared use of drugs? But a thorough search of registration data in key areas of the USA and UK (Stewart, 1992a, 1993a) and of a vast international literature (RootBernstein, 1993) discloses no convincing reports of outbreaks of AIDS in females exempt from riskbehaviour or from circumstances which impose risks upon them.
Use of psychoactive and immunotoxic drugs
An amalgam of debilitating infectious and wasting disease had been noted from the late 1960s onward in young adults and adolescents who injected themselves repeatedly with impure and unsterile, illegallyobtained psychoactive drugs (Gay, 1972; Moss, 1987; Selwyn, 1989). This practice causes bizarre, often intractable infections in the blood and various organs from contaminants in the drugs. Needlesharing, promiscuous sexual intercourse and general disregard of hygiene a notorious feature of the drug scene everywhere leads to sharing also of whatever infections are endemic in that community PCP, HIV, hepatitis, herpes, EBV, VZ and CMV which may impair cellmediated immunity and reverse T4/T8 ratios (Louria,Hensle & Rose, 1967, McDonough, Madden & Falek, 1980; Drew, Mills & Levy, 1985; Creglev & Mark, 1986; Moss, 1987). All the psychoactive drugs currently in use in this way, especially heroin and experimental mixtures, are profoundly depressing to appetite, general health and immunity. Cocaine and crack damage the respitory ephithelium which is a main barrier to all airborne infections. Alternation of excitement and depression leads quickly to habituation, overdosage and reckless disregard of alI the personal and societal consequences of this life style.
Drug use has been escalating in conurbations in the USA for 25 years, and is now the main reason for heterosexual spread of AIDS (Moss, 1987) there and in many other countries. If infection is minimised by using uncontaminated drugs and needles, or especially by opting for less toxic oral drugs such as methadone, many addicts can live equably with their habit for many years (Caper, Goldsmith & Stewart, 1972; Creglav & Mark, 1986). Otherwise, chronic infection, especially with therapyresistant protozoa and fungi, leads to severe disease in target organs and often to death. A pregnant woman in this state transmits her infections and her drugtoxicity congenitally to her child. In some countries, drug addicts donate blood for payment and transmit their latent or active infections to plasma pools. Most or all of the extending range of infections listed as indicators for AIDS (Table 4) and other microflora from the local environment were perceived, together with defects in immunity, in persons in these categories before AIDS appeared. They now include multidrug resistant forms of tuberculosis. The illegal use of drugs is also diversifying as a predictable but usually uncontrollable disaster in large and growing sectors of youth and young adults in the conurbations of the western world. HIV was wellestablished in this population internationally by 1985, and is continuing to spread within it.
Immune system activation
HIV differs from nonretroviral infections because of the affinity between glycoprotein 120 on the surface of the virus and CD4 receptors on Tcells. This facilitates entry of HIV into a minority of cells and initiates a generalized immune response: activation of Tcells, lymphadenopathy, antibodies to envelope proteins, antigenspecific tolerance, neutralization of virus and latency of infection. The HIV hypothesis, postulating reactivation of virus by internal regulation and destruction of immunity by kill ing of Thelper lymphocytes is falsified by the fact that immunity is sufficient to arrest replication of virus and delay onset of further disease, in the absence of risk behaviour, by ten years or more. The usual signal of advance of disease is not viral replication, but a continuing fall in CD4 lymphocytes. In so far as it can occur in other infections, in grafthost disease and in disordered immunity, this is a largely nonspecific event. But it is a significant event in AIDS because it is associated with the appearance of lymphocytotoxic antibodies (LCTAs) acting against nonHLA antigens and peripheral blood B and Tlymphocytes in some haemophiliac and homosexual patients (Pruzanski, Jacobs & Laing, 1983; Kiprov, Anderson & Morand, 1985; Ozturk, Koller & Horsburgh, 1987; Stricker, McHugh & Moody, 1987; Daniel, Schimpf & Opelz, 1989). These occur in seronegative and seropositive patients, but are much more prevalent in the latter and in homosexual and haemophiliac patients with AIDS. They are crossreactive with major histocompatibility (MHC) class II proteins on B and Tcells, with spermatozoa! antigens (Ashida & Schofield, 1987; RootBernstein & Hobbs, 1991) and with antigens from C.allhicans, the cause of the orooesophageal infection which was and is a main and early indicator for AIDS.
Male homosexuals have antispermatozoa! antibodies in the blood, which crossreact with Tcells and have been linked to the occurrence of azoospermia and testicular atrophy in homosexual men (Adams, DonovanBrand & FriedmanKien, 1988; Ma & Armstrong, 1989). The same antibodies have been detected in female patients with AIDS (Sheppard & Ascher, 1990) and are crossreactive with Tcells and HIV antibodies. The common factor is obviously anal intercourse, a main riskfactor for female as for male AIDS. These crossreactions reflect a mixed state of allogenic and autoimmunity in which patients, with and without HIV, reject their own Tcells because they cannot distinguish them from antigens from spermato
zoa, HIV, other infections, foreign proteins and cells in transfusions and in injection needles used for street drugs. This explains the selective incidence of AIDS in homosexual men, in women with bisexual partners or who engage in anal intercourse, and in haemophiliacs, some of whom would be additionally at risk because of innate disorders of immune regulation or of immunosuppression by drugs.
Sheppard and Ascher (1988, 1990, 1992) go further. They see the pathogenesis of AIDS as the outcome of two sets of signals acting on Tcells in a continuous process of immune activation. The first (specific) signal comes from the interaction of a Tcell receptor with an antigenic peptide presented as an MHC molecule following infection with HIV. The second signal is nonspecific and is provided by selfmolecules on cells which react with other Tcell receptors and regulate the activation produced by the first. Most of the progeny of the activated cells ( lymphocytes and thymocytes) are eliminated by 'programmed death' (apoptosis) which restores the immune system to equilibrium (Zacharchuk, Mercep & Chakraborti, 1990), but a minority remain as 'memory' cells in a resting state in which they retain their capacity to respond to an appropriate stimulus (Beverly, 1991). This may come in various ways: in HIV infection, from activation of Tcells by interaction of gp l20 with CD4, leading to lymphadenopathy and nonspecific autoimmune responses. The shift toward programmed cell death then causes a fall in CD4 cells (McClure & Dalgleish, 1992). But there is evidence also of antigen tolerance by clonal deletion of reactive thymocytes and of Bcells of the spleen, by activity of superantigens (Quarantino, Murison & Knyba, 1991) which act on VB regions of Tcell receptors, and by what Sheppard and Ascher (1992) call a 'paradoxicallyintense response' to peptides involved in alloreactivity. It is likely that some of the many drugs used in the treatment of AIDS contribute to this. The outcome depends also on the frequency of exposure to antigens and on the ability of HIV quasispecies to induce the second signal.
One of the wellrecognised immunological anomalies in AIDS (Duesberg, 1989) is that HIV can only replicate in the antigenpresenting Tcells which it suppresses. These cells multiply during the period of general immune activation in the onset of infection, and accept the gpl20/CD4TCR antigen complex (Dalgleish, Wilson & Gompels, 1992), after which they decrease. This is restricted to subsets with MHC Class II allodeterminants which mimic HIV1, supporting the likelihood that the response is autoimmune, similar to that in graftvhost disease, in persons in VB subfamilies with selective HLAassociated susceptibility to HIV1 (Fabio, Scorza & Lazzarin, 1992). In a comprehensive investigation of the immunology, RootBernstein suggests (1992, 1993) that this response is a multiple antigenmediated autoimmunity (MAMA) provoked by the various infections, drugs and alloantigens. These theories offer an explanation of the mechanism of immune activation, but they do not explain the long periods of latency which may follow.
The evidence from this active immunological front presents AIDS in many patients as an autoimmune disease precipitated by rejection of Thelper lymphocytes and thymocytes in complicated crossreactions with any or all of several antigens, and in selfnonself discrimination (von Boehmer & Kisielow, 1990; Sprent, Gao & Webb, 1990) in persons in genetically susceptible subpopulations. This results in an upset of immunological tolerance and a suppression of cellmediated immunity to the point where it can no longer cope with additional infections. With spermatozoa as alloantigens and drugs as independent immunosuppressants included, and with the immunoreactive haplotypes in Caucasian populations defined, this would seem to be sufficient to explain much, if not all, of the pathogenesis as well as the selective and continuing incidence of AIDS almost exclusively in subsets of populations in defined risk groups in North America, Europe and Australasia.
The need for an alternative hypothesis
This has been raised on several occasions especially by Duesberg (1987, 1989), Sonnabend (1989), Evans (1989a) and by the author (1989, 1992a). Sonnabend, working with patients in Manhattan, was the first to explain the vunerability of homosexual men, in particular the effect of spermatozoa in the rectum on immunity. He suggested that risk factors for seroconversion are different from those for AIDS in which autoimmunisation, release of interferon, massive inocula in tranfused blood and bloodproducts, and a trigger effect of coincident viral infections might account for the pathogenesis of ARCs and AIDS.
Conclusion
An alternative hypothesis must explain not only the pathogenesis of immune deficiency in AIDS, but also the pattern of transmission and epidemiology. In the hypothesis presented here, AIDS is presented as a disease acquired in the first place by selfpreferred or imposed behaviours, which in themselves dysregulate immunity and homeostasis while also leading to exposure to various pathogenic and opportunistic infections. The complex syndrome which follows has infectious, immunological and metabolic features. The hypothesis rejects HIV as a unique and sufficient cause of all this but agrees that it is transmissible in sexual secretions and blood, causing HIV disease: lymphadenopathy and febrile illness followed by latency or minimal pathological change during which there is evidence of direct celltocell transmission of virus to migrant mononuclears and neural cells, of direct encephalopathy and of immune activation.
AIDS and AIDSrelated complexes (ARCs) develop, with and without HIV, because heterologous antigens in spermatozoa enter the rectum and bloodstream, or in whole blood and blood concentrates given as transfusions, provoke allogenic responses and elicit antibodies which are toxic to lymphocytes, and cause a fall in CD4 counts. HIV can do the same by joining with CD4 receptors on Thelper lymphocytes presented along with MHC Class II proteins because of molecular affinities. This complex is tolerated, because it is recognisable at first as self, so HIV survives in clones of activated lymphocytes and monocytes in the presence of neutralising antibodies. But repeated infections of the genital, alimentary and respiratory tracts conveyed with various heterologous antigens, as above, maintain the Tcell activation while antilymphocyte antibodies are being formed. This leads to autoimmunity with a fall in CD4 count, reversal of the T4/T8 ratio, energy and programmed cell death of T and Blymphocytes, consistent with the collapse of immunity, and atrophy of thymic and splenic follicles found postmortem in patients dying with AIDS. It explains the general absence of AIDS in immunocompetent persons, the special susceptibility of homosexual men and haemophiliacs, and the risk to the foetus of a mother with AIDS; and it is entirely consistent with the epidemiological pattern of AIDS in the USA and most of Europe to date.
The occurrence of AIDS in drug users is attributable, firstly, to the general immunosuppressive properties of most of the major psychoactive drugs at present in use and secondly, to contaminants and impurities which cause refractory infections and dysregulate immunity. Persons in this risk category often overlap with the male homosexual group. Girls and women place themselves at high risk by taking drugs or by having intercourse with men in high risk groups. If they are pregnant, their infants share these risks by intrauterine or perinatal exposure. Otherwise, the spread of AIDS by heterosexual transmission in either direction is minimal or absent except in subSaharan Africa where registrations are increasing rapidly, but in a totally different clinical and epidemiological pattern which overlaps with other, prevalent infections and with malnutrition.
Predictions made on this basis are accurate to within 10% of registered totals of current and cumulative incidence in the USA and UK. The riskbehaviour hypothesis postulates that, for these reasons, AIDS will continue to occur in persons and communities in defined susceptibility groups although HIV disease will be much more widely prevalent. Along with other organisms (HSV, CMV, VZ, EBV, various protozoa, fungi and bacteria), HIV can be activated from latency by various forms of risk behaviour, as described above, because this leads to an overload of genital, alimentary, pulmonary and systemic infections compounded by dysregulation of natural immunity, either by spermatozoa in the rectum and blood in persons of either sex experiencing traumatic anal intercourse, or from organisms acquired in oral sex, or from the immunotoxic effects of injected or ingested drugs or from selfmedication by broadspectrum antimicrobial agents or, frequently, from al1 of these in lifestyles which disregard elementary rules of hygiene and nutrition. In persons choosing these lifestyles, AIDS is essentially a selfinflicted disease which can only be prevented by awareness and selfcontrol. For persons upon whom these risks are inflicted, one way or another, it is becoming increasingly and tragically obvious that protection is imperative.
Impact of this new hypothesis on research and control of AIDS
The monopolistic hypothesis that HIV I is the unique cause of AIDS has, since 1984, led not only to erroneous predictions, but also to widespread misinformation and grotesque errors in prognosis, treatment, allocation of resources and strategy for research (Rubin, 1988; Adams, 1988; Eigen, 1989; Stewart, 1989, 1992a; Craven, Stewart & Taghavi, 1994). Resources and funds for the longer term are allocated mainly for singlefactor strategy based on the false assumptions (Montagnier, 1994) that a specific vaccine or drug will eliminate or cure AIDS. Even if this were possible, the ethical and logistic problems would be immense. To whom would the vaccine be given? Would recipients be encouraged to continue riskbehaviour? How else would exposure and efficacy be measured? Or will the vaccine or vaccines be used as shotguns on the blind guess that everyone is already at risk? Since heterosexual spread is not occurring in developed countries on anything approaching the scale envisaged in official predictions, it is easy to see how a vaccine used widely at this stage could be given credit for control of a pandemic which is not occurring. On the drug front, the consensus jumped the gun by promoting the use of Azidothymidine (AZT, Zidovudine), a highly cytotoxic drug, for prophylaxis in seropositive pregnant women and infants on the assumption that without it, they would all develop AIDS and die. This policy continues, despite the evidence in the prolonged AngloFrench Trial (Aboulker & Swart, 1993; Concorde,1994) which showed no significant prophylactic effect in symptomfree HIVpositive subjects in terms of survival or disease progression after five years.
If the HIV hypothesis is inadequate or wrong, the risks and misplacement of effort and research since 1984 will be enormous. The alternative hypothesis offered here differentiates HIV infection and disease from AIDS which, in developed countries at least, is a complex amalgam of diseases determined first and foremost by high risk behaviour in subsets of populations in restricted social, ethnic and geographic locations. It postulates that prevention depends essentially upon recognition and control of these existential determinants by education, notification, contact tracing and, if necessary, by legal constraints upon behaviour which places unaware or passive persons, including unborn infants, at equally high or higher risks. The situation in the developing world is even more serious but is different in ways which cannot be understood without a more informative database about the distribution and pattern of AIDS and other lifethreatening and sexuallytransmitted diseases, and about life styles in affected countries.
The data and predictions supporting the alternative riskbehaviour hypothesis are presented here in a manner which opens them in the short term to falsification and correction, for instance, by factual data excluding other diagnoses and confirming the occurrence of destruction of immunity with unremitting signs of AIDS and HIviraemia by secondary transmissions to and between persons not engaging in riskbehaviour, or in infants of seropositive mothers not exposed to direct or indirect risks.*
References
Aboulker, JR & AM Swart, 1993. Preliminary analysis of the Concorde Trial. Lancet 341: 88990.
Acquired Immune Deficiency Syndrome (AIDS) in England & Wales to the end of 1993. 1990. Report of a Working Group convened by the Director of the Public Health Laboratory Service, London, HMSO.
Adams, J.A.D., 1988. AIDS: the HIV myth. London: Macmillans.
Adams, L.E., R. DonovanBrand & A. FriedmanKien, 1988. Sperm and seminal plasma antibodies in AIDS. Clin. Immunol. Immunopathol.46: 442.
Ades, A.E., S. Parker & D. Cubitt, 1992. Two methods for assessing riskfactor composition of the HIVI epidemic in heterosexual women: Southeast England, 19811991. AIDS 6(9): 1031 6.
Advisory Committee on Dangerous Pathogens, January 1990. HIV the causative agent of AIDS and related conditions, 2nd revision; London, HMSO.
Agency for Health and Welfare Services, California. Nov. 8, 1995. AIDS in San Francisco; California Morbidity Reports.
Amer. Assoc. Adv. Sci. Policy Forum (AIDS). 1988. Science 241: 5147.
Anderson, R.M. & R.M. May, 1987. Transmission dynamics of the Human Immune Deficiency Virus (HIV). Nature 326: 137 42,
Ascher, M.S. & H.W. Sheppard, 1988. AIDS as immune system activation: a model for pathogenesis. Clin. Exp. Immunol. 73: 1657.
Ashida, E.R. & F.L. Schofield, 1987. Lymphocyte major histoconr,patability complexencoded class 11 structures may act as sperm receptors. Proc. Nat. Acad. Sci. USA 84: 3395.
Baltimore, D. & M.B. Feinberg, 1989. Editorial: HIV revealed. New Eng. J. Med.321: 1673.
BarreSinoussi, F., I.C. Chermann & E Rey, 1983. Isolation of a Tlymphotropic virus from a patient at risk for AIDS. Science 220: 86870.
Beral, V., D. Bull & S. Darby, 1990. Kaposi's sarcoma and sexual practice associated with faecal contact in homosexual or bisexual men with AIDS. Lancet 339: 6326.
Berkley, S.E, R. WidiWirski & S.l. Odware, 1989. Risk Factors associated with HIV infection in Uganda. J. Inf. Dis. 160: 2230.
Beverly, RC.L., 1991. Is Tcell memory maintained by crossreactive stimulation? Immunol. Today I 1: 203205.
Blattner, W., R.C. Gallo & H. Tenin, 1988. HIV causes AIDS. Science 241: 51413.
Bregman, J. & A. Langmuir, 1990. Farr's Law applied to AIDS projection. JAMA 236: 15225.
Capel, W., B. Goldsmith & G.T. Stewart, 1972. The ageing narcotic addict. J. Gerontol. 27: 10210.
Carlin, R., B.M. Peterlin & D. Derse, 1992. Inhibition of HIVI Tat activity by coexpression of heterologous transactivators. J. Virol. 66(4): 20007.
Carr, E., E. Edmond & R.J. Prescott, 1984. Abnormalities of circulating lymphocyte subsets in haemophiliacs in an AlDSfree population. Lancet 1: 1431 4.
Cheesbro, B., K. Wehrly, Nishio & S. Perryman,1992. Macrophagetropic HIV isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with Tcell tropic isolates. J. Virol. 66(11): 654754.
Concorde: 1993. MRC/ANRS randomised doubleblind controlled trial of immediate and deferred zidovudine in symptomfree HIV infection. Lancet 343: 87181.
Courouce, A., A. Muller & B. Richard, 1986. Falsepositive western blot reactions to HIV I in blood donors. Lancet 2: 5z i 2.
Cox, D.R., R.M. Anderson & H.C. Hiller (ed), 1989. Epidemiological and statistical aspects of the AIDS epidemic. Phil. Trans. S. Soc. Lond. (B) 325: 37187.
Craven, B.M., G.T. Stewart & M. Taghavi, 1994. Amateurs confronting specialists: Expenditure on AIDS in England. J. Pub. Policy 13: 305325.
Creglev, L.L. & H. Mark, 1986. Medical complications of cocaine abuse. New Eng. J. Med.315: 1495.
D'Arminio, M.A., L. Vago & A. Lazzarin, 1992. AlDSdefining diseases of 250 HlVinfected patients: a comparative study of clinical and autopsy diagnosis. AIDS 6: 115964.
Dalgleish, A. & R.A. Weiss, 1984. Prevalence of antibodies to human Tcell lymphotropic virus type 3 in AIDS and AlDSrisk patients in Britain. Lancet 2: 47780.
Dalgleish, A.G., RC.L. Beverley & P.R. Clapham, 1985. The CD4 (T4) antigen is an essential component of the receptors for the AIDS retrovirus. Nature 312: 7637.
Dalgleish, A.G., S. Wilson & S. Gompels, 1992. Tcell receptor variable geneproducts and early HIV infection. Lancet 339: 8246.
Daniel, V., K. Schimpf & G. Opelz, 1989. Lymphocyte antibodies and alloantibodies in HlVpositive haemophilia patients. Clin. Exp. Immunol.75: 178183.
Darby, S.C., C.R. Rizza & R. Doll, 1989. Incidence of AIDS and excess mortality associated with HIV in haemophiliacs in the UK. Report on behalf of the Directors of Haemophiliac Centres in the UK. Brit. Med. J. 298: 10648.
Davey, R.T., L.R. Dayton & J.A. Metcalf, 1992. Indeterminate WB patterns in a cohort at high risk for HIV exposure. J. Clin. Immunol.12(3): 18592
de Biasi, R., A. Rocino & E. Miraglia, 1991. The impact of very high purity Factor VIII concentrate on the immune system of HlVinfected haemophiliacs: a randomized, prospective twoyear comparison with an intermediate purity concentrate. Blood 78: 191922.
De Cock, K.M., B. Soro, I.M. Coulibaly & S.B. Lucas, 1992. Tuberculosis and HIV infection in subSaharan Africa. JAMA 268: 15817.
Dept. of Health and Welsh Office, 1988. Shortterm prediction of HIV infection and AIDS; report of a working group (Chairman: Sir D. Cox), London, HMSO.
Detels, R., R English & B.R. Visscher, 1989. Seroconversion, sexual activity and condom use among 2,915 seronegative men followed for up to two years. J. AIDS 2: 7783.
Drew, W.L., J. Mills & J. Levy, 1985. Cytomegalovirus infection and abnormal Tlymphocyte subset ratios in homosexual men. Ann. Int. Med.103: 613.
Duesberg, RH. & J.R. Schwartz, 1992. Latent viruses and mutated oncongenes: no evidence of pathogenicity. Progress in Nucleic Acid Research. New York: Academic Press.
Duesberg, RH., 1989a. Does HIV cause AIDS? J. AIDS 2: 5145.
Duesberg, RH., 1989b. HIV and AIDS: correlation but not causation. Proc. Nat. Acad. Sci. USA 86: 75562.
Duesberg, RH., 1989c. Response to the AIDS Debate. Naturwissenchafien.
Duesberg, RH., 1987. Retroviruses as carcinogens and pathogens: expectations and reality. Cancer Research 47: 11991220.
Eigen, M., 1989. The AIDS debate. Naturwissenchafien 76: 341540.
Elangovan, B., T. Subramanian & G. Chinnadurai, 1992. Functional comparison of the basic domains of the Tat proteins of HIV types I and 2 in transactivation. J. Virol. 66 (4): 20316.
Evans, A.S., 1989a. Does HIV cause AIDS? An historical perspective. J. AIDS 2: 10713.
Evans, A.S., 1989b. Does HIV cause AIDS? Author's reply. J. AIDS 2: 5157.
Fabio, R. Scorza & A. Lazzarin, 1992. HLAassociated sensitivity to HIV infection. Clin. Exp. Immunol. 87: 1925.
Fauci, A.S., 1988. The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science 239: 61722.
Fordyce, E.J., S. Blum, A. Balanon & R.L. Stoneburner, 1991. A method for estimating HIV transmission rates among female sex partners of male intravenous drug users. Amer. J. Epid.133: 5908.
Gallo, R.C., S.Z. Salahuddin & M. Popovic, 1984. Frequent detection and isolation of cytopathic retroviruses (HTLV III) from patients with AIDS and at risk for AIDS. Science 224: 5003.
Gallo, R.C., 1987. The AIDS Virus. Scientific American 3948.
Gay, G., 1972. Epidemiologic drug trends in San Francisco. Ch 12 in Trends in Epidemiology ed Stewart, GT, Springfield, III: C C Thomas.
Gonlieb, M.S., 1981. Pneumocystis carinii and mucosal candidiasis in previously healthy homosexual men. New. Eng. J. Med.305: 24.
Gunzburg, H.M., P.L. Fleming & K.D. Millar, 1988. Selected public health observations derived from the Multicenter AIDS Cohort Study. J.AIDS 1: 27.
Hanafusa, H., A. Pinter & M.E. Pullman (eds), 1989. Retroviruses and Disease. San Diego, CA, Academic Press.
Hawkes, S., A. Malin, T. Araru & D. Mabey, 1992. HIV infection among heterosexual travellers attending the Hospital for Tropical Diseases, London. Genitourin. Med. 68: 309311.
Hawkins, R.E., L.S. Hickman, S.H. Vermund & M. Carl, 1992. Association of Mycoplasma and HIV infection: detection of amplified Mfermentans DNA in blood. J. Inf. Dis. 165: 5815.
Hilgartner, M.W., 1987. AIDS in Haemophilia. New. Eng. Med. J. 317: 15334.
Ho, D.D., R.J. Pomerantz & J.C. Kaplan, 1987. Pathogenesis of infection with Human Immunodeficiency Virus. New Eng. J. Med. 317: 278285.
Innocenti, R, Ottoman & R Morand, 1992. HIVI in blood monocytes: frequency of detection of proviral DNA using PCR and comparison with the total CD4 count. AIDS Res. Hum. Retroviruses 8: 2618.
Institute of Medicine, 1988. Confronting AIDS: Update. Washington, DC., National Academy of Science 198.
International Classification of Diseases, 10th Ed, 1992. Geneva: WHO.
Jason, J., R.C. Holman & B.L. Evan, 1990. Relationship of partially purified concentrates to immune tests and AIDS. Am. J. Hematol.34: 2629.
Jin, Z., R.R Cleveland & D.B. Kaufman, 1989. Immunodeficiency in patients with haemophilia. J. Allerg. Clin. Immunol.83: 16570.
Katseni, V.L., C.B. Gilroy & B.K. Ryait, 1993. Mycoplasma fermetans in individuals seropositive and seronegative for HIVI. Lancet 341: 2713.
Kiprov, D.D., R.D. Anderson & R Morand, 1985. Antilymphocyte antibodies and seropositivity for retroviruses in groups at high risk for AIDS. N. Eng. Med. J. 312: 1517.
KonoteyAhulu, F.l.D., 1989. What is AIDS? Watford, UK, TenehA'Domeno.
Lancet (Editorial). 1992. Risk of HIV transmission during dental treatment. 340: 1259.
Lemaitre, M., D. Guetard, Y. Henin, L. Montagnier & A. Zerial, 1990. Protective activity of tetracycline analogs against the cytopathic effect of the HlVs in CEM cells. Res. Virol. 141: 516.
Levy, J.A. & J. Chimabukuro, 1985. Recovery of AlDSassociated retrovirus from patients with AIDS and related conditions and healthy persons. J. Inf. Diseases 152: 7347.
Li, P. & C.J. Burrell, 1992. Synthesis of HIVDNA in a celltocell transmission model. AIDS Res. Hum. Retroviruses 8: 2539.
Lo, S.C., R.Y. Wang & PB. Newton, 1989. Fatal infection of silver leaf monkey with a viruslike agent (VLA) derived from a patient with AIDS. Amer. J. Top. Med. Hyg.40: 399 409.
Lo, S.C., 1986. Isolation and identification of a novel virus from patients with AIDS. Amer. J. Trop. Med. Hyg. 35: 6756.
London Declaration on AIDS, 1988. The Times, London, 27 January.
Lourie, D.B., T. Hensle, & J. Rose, 1967. The major complications of heroin addiction. Ann. Int. Med.67: 16.
Louwagie, E, F. McCutchan & G. Van der Groen, 1992. Genetic comparison of HIVI isolates from Africa, Europe and North Amenca. AIDS Res. Hum. Retroviruses 8(8): 41679.
Ludlam, C.A., J. Tucker & C.M. Steel, 1985. HTLV 111 infection in seronegative haemophiliacs after transfusion of Factor Vlll. Lancet 1: 2336.
Ludlam, C.A., 1993. Personal communication.
Ma, P. & D. Armstrong (eds), 1989. AIDS and infections of homosexual men. 2nd Ed. Boston Butterworths.
Makris, M., EE. Preston & D.R. Triger, 1990. CMV infection and progress towards AIDS in haemophiliacs with HIV infection. Lancet 1: 1179.
Mann, J.M., (WHO), 1989. AIDS: a worldwide pandemic. Current Topics on AIDS 2: 110.
Marcus, R., 1988. CDC Needle Stick Surveillance of health care workers exposed to blood from patients infected with HIV. New Eng. Med. J. 319: 111832.
Marmor, M., A.E. FriedmanKien & L. Laubenstein, 1982. Risk factors for Kaposi's sarcoma in homosexual men. Lancet 1: 1083.
Mavligit, G.M., Talpag & F.T. Hsia, 1984. Chronic immunological stimulation by sperm alloantigens. J. Amer. Med. Assoc. 251: 23741.
McClure, M.O. & T.F. Schultz, 1989. Origins of HIV. Brit. Med. J. 289: 12678.
McClure, M.O. & A.C. Dalgleish, 1992. Human immunodeficiency virus and the immunology of infection. Baillere's Clin. Obst. Gynaecol.6: 112.
McDonough, R.J., J.J. Madden & A. Falek, 1980. Alterations in T and null lymphocyte frequency in the peripheral blood of human opiate addicts.J. Immunol. 125:2539.
Medical Research Council, The Edinburgh Haemophiliac Cohort, 1990. Project Report: MRC, London (Update).
Meyer, K.B. & S.G. Panker, 1987. Screening for HIV:L can we afford the falsepositive rate? New Eng. J. Med. 317: 23841.
Midthum, K., G. Garrison & M.L. Clements, 1990. Frequency of indeterminate Western Blot tests in healthy adults at low risk for HIV infection. J. Inf. Dis. 162: 137982.
Mirvish, S.S. & H.W. Haverkos, 1987. Butyl nitrate in the induction of Kaposi's sarcoma in AIDS. New Eng. J. Med. 317: 1103.
Montagnier, L., 12th March, 1994. Quoted by the Times, London.
Morb.Mort.Wkly. Reports.1981;30,250,305,409;and 1982;31, 37,507; and Special Report on Surveillance to 1989. Center for Disease Control, Atlanta, Ga.
Mortimer, RR, 1991.The fallibility of HlV western blot. Lancet37: 2867.
Moss, A.R., D. Osmond & R Bacchetti, 1987. Risk factors for AIDS and seropositivity in homosexual men. Amer. J. Epid. 125: 103547.
Moss, A.R., 1987. AIDS and intravenous drug use: the real heterosexual epidemic. Brit. Med. J. 249: 38990.
New York City, Dept. of Health. 198292. AIDS Surveillance Updates.
Newell, G.R., RW. Mansell & M.R. Spitz, 1985. Volatile nitrites: use and adverse effects in relation to the current epidemic of AIDS. Amer. J. Med. 78: 8116.
Ozturk, G.E., RE Koller & C.R. Horsburgh, 1987. Tbe significance of antilymphocyte antibodies in patients with AIDS and their sexual partners. J. Clin. Immunol. 7: 130 133.
Pagano, M., V. DeGruttola, S. MaWhinney & X.M. Tu, 1991. The HIV Epidemic in New York City; Projecting AIDS Incidence and Prevalence. Report prepared under contract with New York City.
PapadopulosEleopulos, E. & V.F. Turner, 1993.1s the Western Blot proof of HIV infection? Biotechnol. I 1: 696707.
Pfutzner, A., U. Dietrich & U. Von Eichel, 1992. HIVI and HIV2 infections in Bombay, India: evidence for spread of HIV2 and presence of a divergent HIVI subtype. J. AIDS 5 (10): 9727.
Pruzanski, W., H. Jacobs & L.R Laing, 1983. Lymphocytotoxic antibodies against peripheral blood B and Tlymphocytes in homosexuals with AIDS and ARC. AIDS Res.1: 211220.
Public Health Laboratory Service, 1993. Comm. Dis. Rep. 3 (4): 22nd January.
Public Health Laboratory Service and the Communicable Diseases (Scotland) Unit. 198292. AIDS/HIV Quarterly Surveillance Tables.
Quarantino, S., G. Murison & R.E. Knyba, 1991. Human CD4CD8alphabeta+Tcells express a functional Tcell receptor and can be activated by superantigens. J. Immunol. 147: 33193323.
Quinn, C., R Piot & J.B. MacCormick, 1987. Serological and immunological studies in patients with AIDS in North America and Africa. JAMA 257: 261721.
Ragni, M.V., L.A. Kingsley & R Nimorwicz, 1989. HIV heterosexual transmission in haemophilia couples. J. Acq. Immune Defic. Syndrome 2: 55763.
Ragni, M.V., A. Winkelstein & L.A. Kingsley, 1987. 1986 update of HIV seroprevalence, seroconversion and AIDS incidence and immunological correlates of HIV infection in patients with haemophilia. Blood 70: 786 90.
Revision of the CDC surveillance case definition for AIDS, 1987. Morb. Mort. Wkly. Reports 36: supp 35. Center for Disease Control, Atlanta, Ga.
RootBernstein, R.S., 1992. Multiple antigenmediated autoimmunity (MAMA) in AIDS: a possible model for postinfectious autoimmune complications. Res. Immunol.141: 321339.
RootBernstein, R., 1993. Rethinking AIDS. New York: Macmillan.
RootBernstein, R.S. & S.H. Hobbs, 1991. Homologies between Mycoplasma adhesion peptide, CD4 and CLASS II MHC proteins. Res. Immunol.142: 519523.
Rubin, H., 1988. Etiology of AIDS. Science 240: 1389.
Sato, RA., J.A. Chin & J.M. Mann (WHO), 1989. Review of AIDS and HIV infection: global epidemiology and statistics. AIDS 3 (supp) S: 3017.
Schoental, R. 1988. AIDS and neoplasias associated with AIDS. Inter. J. Env. Studies 31: 26978.
Seale, J., 1988. Origin of the AIDS viruses, HIVI and HIV2: fact or fiction? Discussion paper. J. Roy. Soc. Med. 81: 5379.
Seale, J.R., 1987. Pathogenesis and transmission of AIDS. Veterinary Record 120: 4549.
Selwyn, RA., 1989. Issues and the clinical management of IVDU with HIV infection. AIDS 3: suppl 1; 2018.
Seremetis, S., L.M. Aledort & H. Sacks, 1990. Differential effects of CD4 counts of high or intermediate purity Factor VIII concentrates in HIV+ haemophiliacs. Blood 76: 48a and Coagulation Abstracts 1111455.
Serwadda, D., N.K. Sewankambo & J.W. Carswell, 1985. Slim Disease: a new disease in Uganda and its association with HTLVIII infection. Lancet ii: 8502.
Sheppard, H.W. & H. Ascher, 1992. The relationship between AIDS and immunologic tolerance. J. AIDS 5: 1437.
Sheppard, H.W. & M.S. Ascher, 1990. AIDS and programmed cell death. Immunol. Today 12: 423,
Shilts, R., 1987. And the Bank Played On. New York: Penguin Press.
Shioda, T. & J.A. Levy, 1992. Small amino acid changes in the V3 hypervariable region of gp 120 can affect the Tcell line and macrophage tropism of HIVI. Proc. Nat. Acad. Sci. USA 89: 94348.
Sonnabend, J., 1989. Facts and speculation about the cause of AIDS. AIDS Forum 2: 312.
Spencer, L.T., M.T. Ogino, W.M. Danker & S.A. Spector, 1994. Clinical significance of HIV type I phenotypes in infected children. J. Inf. Dis. 169: 4915.
Sprent, J., E.K. Gao & S.R. Webb, 1990. Tcell reactivity to MHC molecules: tolerance v. intolerance. Science 248: 1357 63.
Steel, C.M., D, Beatson & R.J.G. Cuthbert, 1988. HLA haplotype Al B8 DR3 as a risk factor for HlVrelated disease. Lancet 1: 11858.
Stewart, G.T., 19923. Epidemiology and Transmission of AIDS. Soc. Pub. Hlth., Official Handbook 1924, London: Meadowbank Pub.
Stewart, G.T., 1991. AIDS in the UK. Estimates of differences by risk group denominators. AIDS News Supplement, Comm. Dis. Scotland Weekly Report 24: 13.
Stewart, G.T., 1993. AIDS predictions in the UK. Lancet 342:437.
Stewart, G.T., 1990. AIDS. Differences within the UK. AIDS News Supplement, Comm. Dis. Scotland Weekly Report 33: 13.
Stewart, G.T., 1992. Changing casedefinition for AIDS. Lancet 340: 1414.
Stewart, G.T. 1993. Errors in predictions of the incidence and distribution of AIDS. Lancet 342:437.
Stewart, G.T., 1990. Tbe Times, London 24 January.
Stewart, G.T., 1989. Uncertainties aboutAIDS and HIV. Lancet 336: 1325.
Stoneburner, R.L., M.A. Chiasson, I.B. Weisfuse & PA. Thomas 1990. Editorial Review. The epidemic of AIDS and HIV I infection among heterosexuals in New York City. AIDS 4: 99106.
Stricker, R.B., T.M. McHugh & T.J. Moody, 1987. Prevalence of an AlDSrelated autoantibody against CD4+ T cells in humans and monkeys. Blood 70: 127.
Tsoulkas, C., F. Gervais, J. Shuster & R Gold, 1984. Association of HTLV 111 antibodies and cellular immune status of haemophiliacs. New Eng. J. Med.311: 15145.
US Centers for Disease Control, 1993. Revised Classification System for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. Morb Mortal Wkly Rep 1992 41: 119; with Addendum to the Proposed Expansion of the AIDS Surveillance Case Definition. Oct 22, 1992: Atlanta, Ga.
US Dept of Health and Human Services: HIV/AIDS Surveillance 19821992. Centers for Disease Control, Atlanta, Ga.
Vandenbroncke, J.R & V.RA.M. Pardoel, 1989. Autopsy of epidemiological methods: the case of Poppers in the early epidemiology of AIDS. Amer. J. Epid.129: 4557.
Vartamian, JR, A. Meyerhans & H.M. WainHobson, 1992. Highresolution structure of an HIVI quasispecies: identification of novel coding sequences. AIDS 6: 10958.
von Boehmer, H. & R Kisielow, 1990. Self nonself discrimination by Tcells. Science 248: 136973.
WainHobson, S., 1989. HIV genome variability in vivo. AIDS 3: supp 1; 139.
Wallace, R., 1991. Travelling waves of HIV infection on a lowdimensionalsociogeographicnetwork. Soc. Sci. Med.32: 84752.
Ward, J.W., T.J. Bush & H.A. Perkins, 1989. The natural history of transfusionassociated infection with HIV. New Engl. J. Med. 321: 947
Webster, A., C.A. Lee & D.G. Cook, 1989. CMV infection and progress towards AIDS in haemophiliacs with HIV infection. Lancet 2: 6365.
Weiss, R.A. & H.W. Jaffe, 1990. Duesberg, HIV and AIDS. Nature 345: 65470.
Winkelstein, W.W., J.A. Wiley & S. Padian, 1988. The San Francisco mens' study: continued decline in HIV seroconversion rates among homosexualbisexual men. Amer. J. Pub. Hlth.78: 14724.
Witkin, S.S. & J. Sonnabend, 1983. Immune response to spermatozoa in homosexual men. Fertil. Sterlisation 39: 33742.
World Health Organisation, 194892. Weekly epidemiological reports.
Zacharchuck, C.M., M. Mercep & RK. Chakraborti, 1990. Programmed Tlymphocyte death. J. Immunol.145: 40374045.
Zwart, G., J.J. de Jong & T. Wolfs, 1990. Predominance of HIV serotype distinct from LAV l/HTLV 111. Lancet 1: 474.