Chapter 29
AIDS: An Explanation for Its
Occurrence among
Homosexual MenJoseph A. Sonnabend
The following multifactorial model was first proposed in 1983 as an explanation for the occurrence of acquired immunodeficiency syndrome (AIDS) among homosexual men that did not require the participation of a novel infectious agent. Since that time, several important observations have been made that are relevant to the process of disease acquisition then suggested. First are reports directly relating to both the environmental and biologic factors proposed as being important in the development of AIDS among homosexual men. Second, two different human retroviruses have been discovered - human immunodeficiency virus (HIV)-1 and -2 - and are widely perceived as causing AIDS.How does the multifactorial model stand up in light of these new observations? With respect to the HIVs, despite the widespread acceptance of their respective etiologic roles, these must remain conjectural as long as the following two questions (at least) remain open.
The first relates to pathogenesis and asks how H1V-1 and H1V-2 cause AIDS. While a detailed knowledge of pathogenesis is not required in order to attribute an etiologic role to a particular microorganism, the case for H1V-1 as the cause of AIDS rested on two propositions: (1) that HIV directly killed lymphocytes of the CD4 subset; and (2) that HIV is frequently associated with AIDS. Although the mechanism of cell killing remained to be elucidated, it was assumed that HIV was directly responsible because of its tropism for CD4 lymphocytes coupled with the acceptance that the loss of this lymphocyte subset is the hallmark of AIDS. It is now known that insufficient numbers of CD4 lymphocytes are infected to account for their loss by a direct cell killing effect of HIV. Since no mechanism has been demonstrated that would account for the CD4 lymphocyte loss due to a direct cell killing action of the HIVs in vivo, other, less direct mechanisms, including HIV-induced autoimmune mechanisms, have been proposed.
It has also yet to be demonstrated how infection of a small number of CD4 lymphocytes can account for the widespread abnormalities observed in AIDS. It is now known that the tropism of HIV-1 (and presumably that of HIV-2) is not limited to lymphocytes of the CD4 subset. However, infection of B cells and of macrophages by these retroviruses, although demonstrated, has not been shown to contribute to the pathogenesis of AIDS by any mechanism.
Second, there is an alternative explanation to account for the widespread association of HIVs with AIDS that has yet to be excluded. This is that the expression of HIV - a virus that can be maintained in latency-represents an opportunistic reactivation associated with the immune dysregulation resulting from the true cause or causes of AIDS, whatever these may be, and that these causes have been associated with conditions that promote the spread of all infectious agents, pathogenic or not, that can be transmitted by blood or semen. The activation of latent microorganisms, pathogenic or not, is characteristic of AIDS. Thus the expression of HIV is an effect, rather than a cause, of AIDS.
There is no evidence to suggest that carriage of HIV as a provirus, without seroconversion or with seroconversion delayed for years after infection, is not common. If HIV is not the cause of AIDS this might be anticipated to account for the preservation of HIV in nature as well as its frequent association with AIDS. Newer genome detection techniques such as the polymerase chain reaction may indicate that carriage of HIV-1 and HIV-2 is more widespread than the distribution of AIDS (as a disease), or of H1IV seropositivity. A further prediction is that HIV seropositivity found among individuals, such as organ transplant recipients, who are immunocompromised for known reasons will include some whose clinical course is no different from similar but seronegative patients.
The fact that two disparate viruses cause the same disease may not be so remarkable. Their more or less simultaneous emergence into human populations, however, would be a most improbable occurrence. There is no animal reservoir so far shown for HIV-1 or HIV-2. Thus, the likely antiquity of both HIV-1 and HIV-2 must have been associated with their preservation in nature by transmission between humans, vertical, horizontal, or both. This raises the question of why AIDS had not been recognized previously, particularly since, according to current data, FlIV-1 and H1IV-2 have been isolated in geographically distinct areas. The problem, of course, would be compounded if additional HIVs are isolated in yet different geographic areas.
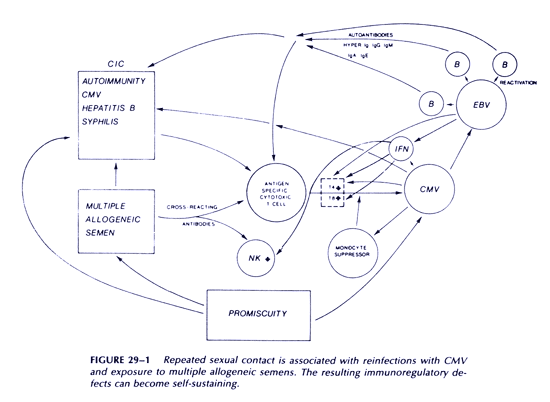
The following model describes a process by which AIDS could have developed in homosexual men that does not require the participation of any HIV or other novel agent. The essential element of this model is that it is an interactive, multifactorial process resulting from repeated exposures, particularly rectally, to large inocula of cytomegalovirus (CMV), together with repeated exposures to multiple alloantigens contained in semen, and repeated exposures to other sexually transmitted pathogens, including Treponema pallidum, resulting in a cumulative impairment of cytotoxic responses against intracellular parasites, including CMV and other herpesviruses. Reactivation of Epstein-Barr virus (EBV) is an important part of the model.
It is a multifactorial model on two levels. It considers the interaction of the individual with multiple environmental factors, and it also describes how the multiple biologic effects generated within the individual by these factors can interact and produce a disease. It takes into account the environmental changes that occurred during the 1970s with respect to sexual lifestyles and the increase in the pool of sexually transmissible microorganisms, pathogenic or not, that was its consequence.
A two-stage process describing the development of AIDS IS presented: an initial stage of disease acquisition, associated with repeated exposures to environmental factors, is followed by a self-perpetuating stage that no longer requires these exposures and has features of a positive feedback system. A role for interferon and possibly tumor necrosis factor in the pathogenesis has now been added.
In summary, this model illustrates how AIDS could have developed in homosexual men as a result of an interaction of known or likely biologic effects generated by repeated exposures to specific infectious and noninfectious environmental factors. Numerous reports now document the specific environmental and biologic features that were regarded as important in the 1983 model, which appears-with minor updating-below.
The occurrence in 1981 of AIDS among a group of homosexual men, predominantly in New York City, San Francisco, and Los Angeles, remains unexplained. Manifestations of the syndrome include opportunistic infections, autoimmunity, and neoplasia. Autoimmunity, once completely ignored as a component of AIDS, now receives much attention.(1, 2) It is a syndrome of multiple diverse manifestations; indeed, this very heterogeneity is one of its essential features.
It had been suggested early that a new and unique transmissible agent was responsible for AIDS, thus linking the disease occurring in homosexual men with a similar syndrome seen among Haitians, intravenous (IV) drug users, and recipients of blood products.(3) This was indeed a serious assertion, and a concern for its far-reaching consequences prompted us to present our model for the genesis of the syndrome in 1983, since it does not require the person-to-person transmission of a new infectious agent. Rather than invoke a single common infectious etiology, this model proposes that different pathways can lead to similar disorders of immune regulation and outlines the mechanisms that may lead to AIDS in homosexual men. A group of patients who closely resemble homosexual men are renal transplant recipients, who experience the same infections, Kaposi's sarcoma (KS), and lymphomas. As is the case with the men with AIDS, renal transplant recipients have an underlying immunologic disorder, but in this instance there is no disagreement that it results from intentional immunosuppressive therapy and the effects of the allograft.
HYPOTHESIS
Any hypothesis regarding the genesis of AIDS must explain why the syndrome has occurred at this time; in short, "Why now?" It is suggested that the new element was an unprecedented level of sexual promiscuity that had developed among a subgroup of homosexual men in New York, San Francisco, Los Angeles, and some other large urban centers since the late 1960s. Homosexual patients with KS and Pneumocystis carinii pneumonia have reported sexual contact with an unusually large number of different partners. This has been a consistent finding in the few epidemiologic surveys that have been reported (4) and will be expanded on in later sections.
We suggest that two distinct stages may be recognized in the development of the syndrome. An initial reversible stage of disease acquisition is followed by a self-sustaining stage of disease progression. It is during the first stage that promiscuity is important, because it is associated with an accumulation of effects that will eventually lead to the second, self-sustaining stage. We believe that the cumulative effects associated with promiscuity result from repeated infection with CMV, reactivation of EBV, and immune responses to spermatozoa, as well as immune responses to alloantigens on all cellular components of semen. A role for interferon in pathogenesis is now also proposed. Each of these will be discussed in some detail.
FACTORS OF PROBABLE ETIOLOGIC IMPORTANCE IN AIDS
CMV and Immunoregulatory Defects
Infection with CMV has several effects on the immune system. There is an activation of T8 suppressor/lytic T cells, with a reduction in the helper/suppressor T cell ratio. These changes resemble those seen in persons with acute EBV infections, but unlike EBV, T subset aberrations may persist for up to> one year following primary infections with CMV.(5, 6) In addition, infection with CMV induces a population of monocytes with suppressor activity.(7) Autoreactive antibodies have been associated with CMV infections, as has the appearance of circulating immune complexes (CICs).(8, 9) Cells infected with CMV as well as other herpes viruses express Fc receptors (10, 11) Additional observation (12) have confirmed and amplified reports on the effects of CMV noted above. CMV can act as a nonspecific polyclonal B cell activator not requiring T cell help.(13) In addition, monocytes infected with CMV in vitro, as well as monocytes isolated from patients with primary CMV mononucleosis, were less able to support mitogen-induced T cell responses.(7, 14, 15) Monocytes infected with CMV release an inhibitor of interleukin-1; this inhibitor is a host cell protein.(12) Moreover, peripheral blood mononuclear cells infected with CMV show a depressed natural killer (NK) cell activity.(16, 17)
The suggestion that CMV infection contributes to the immunologic perturbation in AIDS has now received support from at least two studies. Detels et al. noted a relationship between CMV antibody titer and T cell subset abnormalities and evidence for the acquisition of CMV infection through receptive anal intercourse.(18) Drew et al. also provide evidence for an effect of CMV infection on T cell subsets in homosexual men.(19) The recent demonstration that CMV contains a protein homologous to major histocompatibility complex (MHC) class I antigens presents another possible mechanism for an immunosuppressive effect of CMV.(20)
The following points are relevant to an association between CMV infections and sexual promiscuity:
1. CMV is excreted in saliva, urine, and semen. Viral titers are probably highest in semen.(21)
2. Asymptomatic carriage of CMV in semen may persist for over one year.(22)
3. CMV antibody has been detected in 94% of homosexual and 54% of heterosexual men attending a venereal disease (VD) clinic. The IgM isotype was detected in 57% of homosexual men, compared with 4% of heterosexual men.(23, 24)
4. The prevalence of CMV viruria among homosexual men attending a VD clinic was 7% to 14%. In this study it was pointed out that the excretion rate would probably have been higher had semen been sampled.(23) It would probably also have been higher had highly promiscuous populations been selected for study.
5. Reinfection with CMV can occur. It is possible to show that a single individual may be infected with more than one strain of CMV by comparing nucleic acid fragments from different virus isolates.(24) Drew and Huang have now shown that four AIDS patients had at least two different CMV isolates from their organ cultures at autopsy.(25)
The frequency with which an individual will be reinfected with CMV is a function of both the number of different sexual contacts as well as the prevalence of CMV carriage in the population with whose members the individual interacts. We suggested that conditions had become such, at least in New York City, during the prior ten years that the prevalence of CMV carriage in populations of highly promiscuous men was at least 10% and may well have been higher.
The high rate of CMV carriage in homosexual men has been further documented in San Francisco, North Carolina, and New York State.(26, 27) The carriage of CMV in semen among sexually active homosexual men in New York City, in fact, reached 40% in 1983 (Lange M, personal communication, 1986). The carriage of CMV in semen, with repeated rectal infection with high-titered inocula, is important to this model.
Reactivation of EBV
Almost all adults will have become infected with EBV, which remains latent in B cells following primary infection. EBV infects B cells, which possess receptors for the virus,(28) and has the capacity to activate B cells to immunoglobulin synthesis. EBV is thus a polyclonal activator and can act as such in the absence of T cell help.(29, 30) This point is significant, since many men with AIDS show evidence of polyclonal B cell activation, and this is seen despite the virtual absence of T helper cells in some of the patients.(31) About one-third of B cells exposed in vitro can be infected by EBV, and about 10% of infected cells will be activated to immunoglobulin synthesis.(32) Among the mechanisms that have evolved to deal with this B cell infection, NK cell activity is important.(33) In addition, suppressor T cells (with a surface phenotype defined by a TX monoclonal antibody) are activated and play a role in containing primary infections by suppressing B cell activation and proliferation.(33) In seropositive individuals, a different type of cytotoxic T cell is rapidly activated. Unlike the suppressor/lytic T cell evoked curing a primary infection, these T cells (memory T cells) from seropositive individuals are specific for EBV-infected B cells.(34) These two types of T cells also differ in the kinetics of suppression of B cell activation to immunoglobulin synthesis.(34) The viral antigen-specific T cell is also HLA restricted, but while T8 cytotoxic cells recognize viral antigens on the surface of the infected cell in the context of class I MHC products, cytotoxic T cells with a T4 surface phenotype recognize antigens in the context of class II MHC products(35) During many viral infections, Hl.A-restricted antigen-specific cytoto3xic T cells are generated.(36)
We propose that, because of their immunosuppressive effects, CMV and possibly some other viruses cause repeated episodes of EBV reactivation. Multiple herpesvirus infections have been noted,(37) and reactivation of EBV has also been seen in some other states of immunodeficiency not directly resulting from viral infections. Administration of cyclosporin A, for example, has been associated with reactivation of EBV.(38, 39) Among agents that induce EBV in vitro are corticosteroids.(40) In 1983 we found that the majority of 50 homosexual men examined showed EBV reactivation patterns (Purtilo D, Sonnabend J, unpublished data). Often, patients with AIDS develop chronic lymphadenopathy and other features of chronic infectious mononucleosis.(41)
Numerous reports now document that EBV reactivation is a common feature in homosexual men with, and at risk for, AIDS. EBV genome copies were detected in lymph node specimens from homosexual men with lymphadenopathy, (42) including those who did not demonstrate an EBV reactivation pattern, in that antibodies to EBV early antigens were absent.
Defective T cell regulation of EBV-infected B cells in AIDS was demonstrated by Birx et al. (43) and was noted and reported by us in 1983.(42)
Chang et al. (44) noted an increase in the number of EBV-infected B cells in homosexual men with lymphadenopathy. An enhanced antibody response to a broad spectrum of EBV antigens was noted by Sumaya et al., (45) resembling that seen in reactivated EBV infections. These authors also confirmed the frequent presence of IgA anti-VCA (viral capsid antigen) antibodies we reported previously.(42)
Further evidence for EBV reactivation in AIDS-related complex (ARC) patients was provided by Ragona et al., (46) who also demonstrated an impairment of specific anti-EBV cytotoxic responses. Asymptomatic homosexual men underwent frequent reactivation or reinfection with EBV.(47) Men who were HIV-reactive demonstrated even higher anti-EBV VCA IgG titers.
The suggestion was made that EBV may be reactivated by HIV; however, the converse could also be true, or both viruses could be reactivated by the same circumstances.
It has thus been ampfy demonstrated that T cell control of EBV-infected B cells is defective in AIDS patients and that EBV reactivation is frequent in AIDS and AIDS-associated conditions.
The resemblance of AIDS patients to renal transplant recipients has been mentioned. It is of great interest that in renal transplant recipients, specific T cell immunity to EBV is impaired, (48) and the lymphomas that they develop contain the EBV genome.(49) The EBV genome has now been detected in AIDS-associated lymphomas.(50)
With successive bouts of EBV reactivation, increasing numbers of B cells will be infected, some will be driven to immunoglobulin synthesis, and a variety of antibodies, possibly including some autoantibodies, (30) will be produced. Many patients show evidence of enhanced immunoglobulin synthesis, involving IgG, IgA, IgM, (51) and even IgE isotypes (Wallace J, personal communication, 1982), despite diminished T helper function. The T cell independent, polyclonal activation of B cells by EBV could explain this paradox.
The hyperimmunoglobulinemia associated with AIDS is now well documented. IgA and IgG are more frequently elevated than IgM. Increased IgD levels have also now been documented.(52) Immunoglobulin elevations may be one of the earliest AIDS-associated abnormalities demonstrable in asymptomatic homosexual men. As observed by Zolla-Pazner, (2) the hyperimmunoglobulinemia in asymptomatic homosexual men may result in part from multiple and repeated sexually transmitted infections.
A study of homosexual men selected for HIV seropositivity indicated that IgA elevations were predictive of a subsequent decline of T4 T lymphocyte numbers.(53) Our own studies have indicated an inverse correlation between T4 lymphocyte numbers and IgA levels, while IgG levels showed a positive correlation with the T8 lymphocyte subset.(54)
Polyclonal Activation of B Cells and Autoimmunity
Many AIDS patients show evidence of autoimmunity. Our finding in 1983 of positive antinuclear antibody responses in AIDS patients has been confirmed, (54) is has the occasional presence of rheumatoid factor. Antibodies reactive with T cells have also been frequently reported and are discussed later in the chapter. in antiplatelet antibody in homosexual men with idiopathic thrombocytopenic purpura (ITP) has been described.(55) IgG anti-IgG F(ab')2 antibodies have also been described in patients with AIDS or at risk for developing AIDS.(56) Auto-antibodies against platelets and granulocytes were also reported by Van der Lelie et al.(57)
It has been recently proposed that autoimmunity in AIDS is induced by HIV infection, as a mechanism to explain the T cell loss in the absence of a clear-cut, direct, in vivo cytocidal effect of HIV. For example, Andrieu et al.(58) propose that because of a molecular mimicry between the HIV envelope protein and class 11 MHC antigens, the immune response against HIV becomes an autoimmune response against class II MHC antigens. Ziegler and Stites propose a similar autoimmune response directed at MHC class II antigens.(59) Another mechanism suggested is that free gp 120 may attach to the T4 molecule on the lymphocyte and thus present a target for antibody-dependent cytotoxic responses. There is, however, no evidence for the presence of such a mechanism in AIDS patients.
The above authors relate the development of anti-T cell autoimmunity to HIV infection. In contrast, our model proposes that anti-T cell antibodies appear as the result of multiple alloimmunization and, to some extent, as part of the polyclonal B cell activation.
It has been reported that spermatozoa express a T4 type of structure.(60) Thus, rectal insemination could induce antibodies reactive with T4 molecules as a result of exposure to spermatozoa, as well as to other cells in semen.
The best documented clinical evidence of autoimmunity is a thrombocytopenia associated with anti-platelet antibodies.(61) It is likely that the leukopenia, and some unexplained rashes frequently observed in these patients also result, at least in part, from autoimmunity. Antinuclear antibody (ANA) was found in 2 of 37 homosexual men with AIDS at a titer of 1:100, and two-thirds of these men had ANA titers of 1:10; 3 of 37 had elevated titers to> (ssDNA), 4 of 37 exhibited rheumatoid factor, and 13 of 37 had circulating immune complexes by the CIq binding assay (Sonnabend J, first edition). Cryoglobulins are detectable in serum during the course of infectious mononucleosis, (62) and we would predict their presence in AIDS. An unusual acid-labile form of alpha interferon has been detected in the sera of many homosexual AIDS patients.(63-65) This type of interferon has been found in systemic lupus erythematosus and some other autoimmune diseases. Its presence in AIDS is further evidence for an autoimmune component in this disease. It is likely that additional clinical manifestations of autoimmunity will become apparent as observations are extended.
Interferon
We propose that the sustained presence of high levels of interferon p1ays a ro1e in the pathogenesis of AIDS. The appearance of interferon in the sera of patients with AIDS-related conditions has been shown to carry an adverse prognostic significance for the development of the full-blown syndrome.(66, 67) The AIDS-associated acid-labile alpha interferon is similar to that which appears in the sera of patients with autoimmune diseases such as SLE.(68-71) There is evidence from animal model systems that interferon may indeed contribute to the pathogenesis of disease in SLE.(72, 73)
The following observations suggest that the sustained presence of high levels of interferon may contribute to the pathogenesis of AIDS:
1. Interferon selectively inhibits the T4 lymphocyte subset m vitro while exerting a slight stimulatory effect on the T8 subset.(74)
2. Interferon can activate T suppressor cells to produce a soluble immune response suppressor that may inhibit antigen-presenting macrophages.(75)
3. Interferon suppresses the proliferative response of lymphocytes to mitogens and alloantigens.(76)
4. Administration of interferon results in lymphopenia, granulocytopenia, and thrombocytopenia.(77)
5. Interferon may also inhibit lipoprotien lipase and elevate serum triglycerides and depress serum cholesterol. These changes are characteristic in AIDS. Such changes can also be induced by tumor necrosis factor or cachectin. Tumor necrosis factor levels are elevated in the sera of AIDS patients.(78)
Interferon also affects immediate hypersensitivity reactions by enhancing the release of histamine from basophilis, (77) thus contributing to drug hypersensitivity and the unexplained rashes common in AIDS. Exacerbations of psoriasis, also common in AIDS, have been associated with the presence of circulating interferon.
Although interferon boosts NK cell activity m short-term exposure, prolonged treatment with interferon actually depresses NK activity.(79) Indeed, incubation of peripheral blood mononuclear cells (PBMCs) from patients with AIDS with alpha-2 interferon did not result in the enhancement of NK activity that was seen with PBMCs from healthy donors.(80) This effect could result from the fact that elevated levels of circulating alpha interferon rendered NK cells unresponsive to in vitro incubation with interferon.
Interferon increases endonuclease L activity in treated cells. On prolonged exposure to interferon, however, this enzymatic activity declines. This may ace count for the low endonuclease level in the PBMCs of AIDS patients.(81) The decline of endonuclease activity may be an adaptive response to prolonged exposure to interferon as may be the down regulation of interferon receptors. Interferon's antiviral activity may therefore not be fully expressed, and its toxicity may also be limited by these adaptive responses in diseases such as AIDS, which are characterized by the sustained presence of high levels of circulating interferon. In vivo correlations have shown that high interferon levels are associated with low T4 cell levels and, interestingly, with high IgA levels as well.(82)
In vivo correlations have shown that high interferon levels are associated with low T4 cell levels and, interestingly, with high IgA levels as well.(82) An increase in IgA levels appears to be an adverse prognostic marker.(83)
Finally, abnormal inclusions noted in the T lymphocytes of AIDS patients on electron microscopy can also be induced by incubating healthy lymphocytes with alpha interferon in vitro.(83)
Immune Responses to Semen
It was of interest to ask if exposure of men to multiple allogeneic semens can induce deleterious immune responses. Witkin and Sonnabend studied immune responses to spermatozoa in 18 homosexual men. Antisperm antibodies of IgG and IgA isotypes were found in 10 and 2 of the 18 men, respectively. Circulating immune complexes were elevated in two-thirds of the men, and sperm-related antigen was found in the sera of some.(84) Semen is immunogenic when deposited in the rectum.(84) Antisperm antibodies could be induced in rabbits following the careful, atraumatic introduction of pooled rabbit semen into the rectum.(85) Thus one possible factor contributing to immunologic impairment could be CICs associated with sperm-related antigens. There are antigens expressed on cells in the ejaculate, including HLA antigens and gangliosides that are shared by lymphocytes.(86) For example, spermatozoa express a ganglioside antigen, asialo GM,, which is also present on NK cells.(87) We have now shown that antibodies to asialo GM' are indeed present in AIDS patients.(88) Many AIDS patients show diminished NK function.(51) Sperm-induced allogeneic immunization was associated with immune dysregulation in individuals who were anal sperm recipients.(89) In addition to the deleterious effects induced by the immune response to the components of semen, direct immunosuppressive effects of semen are well recognized.(90) It is thus possible that repeated exposure to different allogeneic semens may eventually lead to the appearance. of antibodies autoreactive with T lymphocytes and NK cells. It is predictable that multiple anti-HLA antibodies will be found in promiscuous homosexual men who have never received blood transfusions. The diversity of the anti-HLA antibodies may in fact provide an objective measure of promiscuity. The fact that AIDS appears to be of only recent occurrence in homosexual men argues that exposure to allogeneic semen cannot in itself cause substantial morbidity. We propose that immune responses to semen may provide a background of immune suppression, not only promoting repeated CMV infections, but also exacerbating the resulting immunologic disorders.
Circulating Immune Complexes in AIDS
CICs have been detected in many patients with AIDS. There are now numerous reports of CICs in AIDS patients as well as in healthy homosexual men. McDougal et al. (91) showed a correlation with CICs and depressed T4 lymphocyte counts. Undoubtedly, CICs are very heterogeneous with respect to the antigenic component, and there is as yet no proof that any contribute to the development of the immune dysregulation characteristic of AIDS. In some patients, CICs may contribute to thrombocytopenia, polyserositis, arthritis, peripheral neuropathy, and nephropathy. We suggest that CICs may also contribute to the underlying immune disorder. The expression of erythrocyte C3b receptors is impaired in AIDS.(92-94) This is an important component of the mechanism for clearing CICs.
As mentioned earlier, herpesvirus infected cells may be induced to express Fc receptors.(10, 11) This is of potential importance in a host with high levels of CICs. One possible mechanism by which this phenomenon might contribute to pathogenesis is that binding of CICs to Fc receptors could interfere with target recognition by cytotoxic lymphocytes.
Our additional observations have shown a clear correlation between promiscuity and the presence of CICs: 13 of 13 homosexual patients with Kaposi's sarcoma and 6 of 10 promiscuous homosexual men had CICs, whereas CICs were present in only one of eight nonpromiscuous homosexual men (Witkin S, Safai B, Krim M, Sonnabend J, unpublished observations). Undoubtedly, many different antigens participate in immune complex formation in these men. Hepatitis B, syphilis, and CMV are among the infections that are highly prevalent in these men and that can be associated with immune complexes. The association of CICs with syphilis is well documented, as is a depression of NK cell function.(95, 96) Additional contributions to the CICs may appear once autoantibodies are produced. A further contribution is from sperm-related antigens, and indeed their presence in CICs in promiscuous homosexual men who have antibodies to spermatozoa has already been demonstrated.(84)
MECHANISMS OF DISEASE ACQUISITION AND TRANSITION TO A SELF-SUSTAINING STAGE
We propose that the first stage of disease acquisition is a period of frequent sexual contact with different partners in a setting in which the prevalence of CMV carriage is such that repeated infection>n with this virus will occur. These repeated infections arc associated with an accumulation of effects that, in aggregate, eventually result in a switch to a self-sustaining condition characterized by an inability of cytotoxic lymphocytes to clear CMV infected cells. Antigen-specific cytotoxic T cells against CMV-infected targets have been shown to be functionally defective in AIDS.(97) The critical concept during the initial stage is that of a cumulative process involving the following:
1. An increasing level of CICs, which may react with Fc or complement receptors on some T lymphocytes and interfere with their cytotoxic function. Herpesvirus-infected cells, including CMV-infected cells, express Fc receptors and thus may bind CICs and block target recognition by cytotoxic lymphocytes.
2. The appearance in increasing concentrations of antibodies that are crossreactive with cytotoxic T cells and NK cells. The specific targets may be regulatory or effector T cells. The consequence is impaired cytotoxicity. Antibodies reactive with T lymphocytes and NK cells may result from polyclonal B cell activation or from immunization by cross-reactive antigens present in the ejaculate.(98-101) Anti-T cell antibodies have now been repeatedly described in AIDS.
3. A diminishing ratio of T4 helper to T8 suppressor cells. The action of cytotoxic T lymphocytes would be susceptible to T8 suppression. CMV and EBV infections, as well as toxoplasmosis (which is not uncommon in AIDS), have been associated with T subset aberrations. These changes are evoked by antigens expressed on the surface of the infected cell. Persistence of infection will maintain these subset changes.
These three general influences-autoantibodies, CICs, and a decrease in T4:T8 subset ratioconspire to inhibit an effective cytotoxic response to CMV-infected cells. The relative contribution to each might vary from patient to patient.
Eventually, the immunosuppression becomes irreversible and self-sustaining, and independent of promiscuous sexual behavior. The sustained immunoregulatory disorders impair cytotoxic responses to other intracellular parasites, which are responsible for opportunistic infections.
Figure 1 summarizes the mechanism of self-perpetuation in this disease; the essential feature of this second stage is an inability to mount an effective cytotoxic immune response against CMV-infected cells. This second stage has features typical of positive feedback systems.
DISCUSSION
Our model may well be less important as a representation of actual disease mechanisms than as a conceptual framework useful in formulating approaches to research on disease mechanisms and strategies toward rational intervention. In contrast to diseases resulting from infection with a single agent, this model proposes that a disease can result from sustained or repeated exposure to several different infectious and noninfectious agents that alone, as single exposures, are not associated with significant morbidity. Disease develops from the combined and cumulative effects of sustained or repeated exposure to multiple factors rather than following an incubation period after infection with a single agent.
As discussed in another presentation of this model, (104) the dispersal of the elements of the immune system, the variety of different specific and nonspecific effector and regulatory functions, and the chemical diversity of the short- and long-range signals employed imply a great number and variety of vulnerable targets and therefore a susceptibility to many different influences. This model illustrates how the interaction of known or likely effects of specific environmental exposures can lead to the development of progressive immune disregulation in homosexual men repeatedly exposed to the environmental factors in question.
Many factors have been shown to have an adverse effect on immune function. If their interaction can produce disease, then we should expect to encounter more clinical immune disfunction in environments in which these factors are present in greater concentrations. One such environment was homosexual bathhouses in large urban settings in the late 1970s. Similarly, the sharing of needles by many IV-drug users provides the opportunity for frequent exposure to immunosuppressive factors. In Africa, malnutrition coupled with repeated protozoan infections constitutes an immunosuppressive burden.
As also previously discussed, (104) this multifactorial model lends itself to a formal epidemiologic analysis, which is true at two levels. First, we must have a better understanding of the environments in which AIDS develops and the ways in which affected individuals have interacted with those environments. Second, the analysis of the interactions of the various biologic effects generated by these exposures is also an appropriate and important epidemiologic undertaking.
We are aware of the conjectural nature of important aspects of this model. However, corroboration can he readily sought. For example, one can compare CMV excretion rates among different populations distinguished by different levels of promiscuity and sexual preferences and correlate these rates with the prevalence of AIDS. Perhaps the behavioral and cultural aspects that appear to be associated with the genesis of AIDS are the most troublesome; they are also critical, because they suggest an explanation for the occurrence of the syndrome at a particular time and location. Here, too, it should be possible to document whether significant changes in patterns of sexual behavior occurred in New York City in the 1970s.
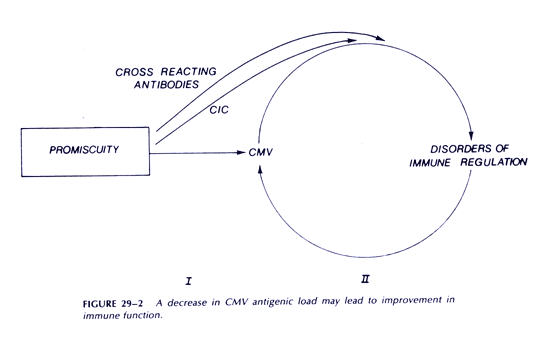
Our model suggests some approaches to patient management that are of immediate practical importance. Both humoral (autoantibodies and CICs) and cellular (inversion of T4:T8 ratios and depressed NK cell activity) factors impair antiviral cytotoxic responses. Methods to remove humoral factors including interferon, such as plasmapheresis, may deserve serious consideration. There are other examples of potentially useful intervention. Cyclophosphamide may control increased immunoglobulin production, and in low dose may have an additional beneficial effect, since it preferentially inhibits T8 suppressor cells. Appropriate monoclonal antibodies may also selectively remove T8 suppressor cells. This subset also includes cytotoxic T lymphocytes, so some obvious caution is required in such an approach. These are examples of approaches to improving cytotoxic function. Any such improvement may set in motion a process leading to recovery. The hope is that some reduction in CMV antigenic load will itself lead to further improvement in immune function (Figure 2). Clearly, it is important to develop and test effective treatments for CMV and EBV infections.
ADDITIONAL COMMENTS ON THE QUESTION OF A SPECIFIC AGENT AS THE CAUSE OF AIDS
Traditionally, social comment in the context of a scientific communication has been regarded as inappropriate. However, in this instance, the potential for adverse social effects of a particular scientific proposal appeared so great that we believe it justified to abandon the traditional restraint on social comment. In short, if groups that already bear a heavy burden of stigmatization are perceived to carry a lethal virus capable of spreading to and decimating the population at large, the danger of consequent brutalization of such groups is only too real. This situation is even more perilous to the groups in question if there is a test that reportedly can identify apparently healthy individuals who belong to these groups, who carry the putatively lethal virus.
Because of the potential for the abuse of individuals identified as a source of contagion, it is especially important to make the distinction between hypothesis and scientific fact. Few would question the inappropriateness of creating public policy on the basis of mere conjecture. Unfortunately, in the case of AIDS such a distinction has not been made.
In addition to the social consequences, the acceptance as fact that HIV-1 and HIV-2 cause AIDS has had the following consequences:
1. Research on other etiologic factors has not been pursued.
2. Aspects of pathogenesis apparently unrelated to HIV have not been investigated. The roles of CMV and EBV infections and of sustained exposure to high levels of interferon as factors contributing to the underlying immune disregulation have yet to be explored.
3. Treatment models other than antiretroviral approaches have not been developed.
4. Patient management strategies have yet to be addressed. This issue has been virtually ignored in the belief than an effective antiretroviral approach will make these considerations redundant.
This model has attempted to describe the development of AIDS as a response to sustained or repeated environmental insults to the immune system. A twostage mechanism of disease acquisition, the second stage having features of a positive feedback system, has been described. The details in this model have been confined to the development of AIDS in homosexual men. Analogous models can be developed for other groups.
Although there can be little doubt that AIDS is a new phenomenon, at least in its epidemic form, among homosexual men, this cannot be said with confidence for any of the other groups. In any group, unless suspected, P. carinii pneumonia would not have been detected because its diagnosis required an open lung biopsy (before 1982).
The consequences of an impaired immune response may be similar, although the pathways that lead to it can be diverse. The route that we believe leads to immunosuppression in one group of patients has been the subject of this chapter. This model has also been presented elsewhere.(103-105)
The above paper was published as chapter 29 in "AIDS and Infections of Homosexual Men", edited by P. Ma and D. Armstrong, (2nd edition, 1989)
This work was supported in part by National Institutes of Health grants HD16586 and HD16587 (to Steven S. Witkin, Ph.D) and CA30196-01, American Cancer Society Grant RD- 161, the Nebraska State Cigarette Tax LB 506, and the Lymphoproliferative Research Fund (to David T. Purtilo, M.D.). This presentation would not have been possible without the critical participation and support provided by Mathilde Krim, Ph.D. I am indebted to her. I am also grateful to Craig Metroka, M.D., for many useful discussions. In addition, I would like to express my appreciation to the following individuals for helping me in this work and in preparing the manuscript: Lillian Waldmann, Anne Marie Bongiovanni, Michael Jurgielski, Paul Krueger, John Donley, Terry Fonville, Harley Hackett, and Suzanne Phillips. My patients have actively participated in the studies, and I acknowledge their role as collaborators.
REFERENCES
1. Zolla-Pazner S. B Cells in the pathogenesis of AIDS. Immunol Today 1984;5:289.
2. Zolla-Pazner S. Serology. In: Ebbesen P, Biggar RJ, Mel bye M, eds. AIDS. Copenhagen: Munksgaard, 1984;1SI.
3. Centers for Disease Control. Aequired immune deficiency syndrome (AIDS): Precautions for clinical and 1aboratory staffs. MMWR 1982;31:S77.
4. Marmor M, Laubenstein L, William DC, et se. Risk factors for Kaposi's sarcoma in homosexua1 men. Lancet 1982;1:1083.
5. Carney WP, Rubin RH, Hoffman RA, et al. Analysis of T cell subsets in cytomegalovirus mononuc1eosis. J Immunol 1981;126:2114.
6. Reinherz EL, O'Brien C, Rosenthal P, Schlossman SF. The cellular basis for viral induced immunodeficiency: Analysis by monoclonal antibodies. J Immunol 1980;125:1269.
7. Carney WP, Hirsch MS. Melchanisms of immunosuppression in cytomegalovirus mononucleosis: 11. Virus-monocyte interaCtioJns. J Infect Dis 1981;144:47.
8. Kantor GL, Goldbey LS, Johnson Bl. Immunological abnormalities induced by postperfusion cytomegalovirus infection. Ann Intern Med 1970;73:553.
9. Olding LB, Kingsburg DT, Oldstone MBA. Pathogenesis of cytomegalovirus infection. Distribution of vira1 products, immune complexes and autoimmunity during 1atent murine infection. J Gen Viro1 1976;33:267.
10. Keller R, Peichel R, Goldman JN. An IgG-Fc receptor induced in cytomegalovirusinfected human fibroblasts. J Immunol 1976;116:772.
11. Rahman AA, Teschner M, Sethi KK, Brandis HE. Appearance of IgG Fc receptor(s) on cultured human fibroblasts infected with human cytomegalovirus. J Immunol 1976; 117:253.
12. Sissons JG. The immunology of cytomegalovirus infection. J R Coll Physicians Lond 1986;20:40.
13. Hutt-Fletcher LM, Balachandran N, Elkins M. B Cell activation by cytomegalovirus. J Exp Med 1983;1S8:2171.
14. Rinaldo CR, Black PH, Hirsh MS. Interactions of virus with mononucleosis due to cytomegalovirus. J Infect Dis 1977;136:667.
15. Rinaldo CR Jr., Carney Wl,, Richter BS, et al. Mechanisms of immunosupprcssion in cytomegaloviral mononucleosis. J Infect Dis 19XO;141:488.
16. Rice GP, Schrier RD, Oldstone MB. Cytomegalovirus infects human lymphocytes and monocytes: virus expression is restricted to immediate-early gene products. Proc Natl Acad Sci USA 19X4 j81:6134.
17. Schrier RD, Rice GP, Oldstone MB. Suppression of natural killer cell activity and T cell proliferation by fresh isolates of human cytomegalovirus. J Infect Dis 1986;153:1084.
18. Dete1s R, Visscher BR, I.ahey JL, et al. The relation of cytomegalovirus and Epstein-Barr virus antibodies to T ce1l subsets in homosexually active men. JAMA 1984;251:1719.
19. Drew WL, Mills J, Levy J. Cytomegalovirus infection and abnormal T Iymphocyte subset ratios in homosexua1 men. Ann Intern Med 1985;103:61.
20. Beck S, Barre1l B. Human cytomegalovirus encodes a glycoprotein homologous ta MHC class I antigens. Nature 1988;331:269.
21. Lang D, Kummer JF. Demonstration of cytomegalovirus in semen. N Engl J Med 1972;287:756.
22. Lang DJ, Kummer JF, Hartley DP. Cytomegalovirus in semen: Persistence and demonstration in extracellular fluids. N Engl J Med 1974;291:121.
23. Drew WL, Lawrence, Mintz L, Miner RC, et al. Prevalence of cytomegalovirus infection in homosexual men. J Infect Dis 1981;143:188.
24. Drew WL, Miner RC, Ziegler J, et al. Cytomegalovirus and Kaposi's sarcoma in young homosexual men. Lancet 1982;2:125.
25. Drew WL, Huang E. Etio1ogy: role of cytomegalovirus. In: Ziegler JL, Dorfman R, eds. Kaposi's sarcoma. New York: Marccl Dekker, Inc., 198 X; 113.
26. Mintz L, Drew WL, Miner RC, et al. Cytomegalovirus infections in homosexual men. An epidemiological study. Ann Intern Med 1983;99:326.
27. Buimovici-Klein E, Lange M, Ong KR, et al. Virus isolation and immune studies in a cohort of homosexual men. J Med Virol 1988;25:371.
28. Jonda1 M, K1ein G. Surface markers on human T and B cells. Vl. Presence of Epstein-Barr virus receptors on human B Iymphocytes. J Exp Med 1973;138:137.
29. Rosen A, Gerge1y P, Jondal M, Klein G. Polyclonal Ig production after EpsteinBarr virus infection of human leukocytes in vitro. Nature 1977;267:52.
30. Fong S, Vaughan JH, Tsoukas CD, et al. Selective induction of autoantibody secretion in human bone marrow by Epstein-Barr virus. J Immunol 1982;129:1941.
31. Gottlieb MS, Schrott R, Schanker HM, et al. Pneumocystis carinii pneumonia and mucosa1 candidiasis in previous1y healthy homosexual men. N Engl J Med 1981;305:1425.
32. Bird AG, Britton S, Ernberg 1, Nilsson K. C.haracteristics of Epstein-Barr virus activation of human B Iymphocytes. J Exp Med 1981;154:832.
33. Purtilo DT, Sakamoto K. Epstein-Barr virus and human disease: Immune respunsec determine the clinical and pathologic expression. Hum Pathol 1981;12:677.
34. Tosato GG, Magrath IT, 131acsc RM. T cell-mediated immunoregulation of Epstein-Barr virus (EBV)-induced B lymphocyte activation in EBV seropositive and EBV seronegative individuals. J Immunol 1982;128:575.
35. Meuer SC, Sch1ossman SF, Reinherz, EL. Clonal analysis of human cytotoxic T lymphocytes: T4 + and T8 + effector T ce11s recognize products of different major histocompatibi1ity comp1ex regions. Proc Nat1 Acad Sci USA 1982;79:4590.
36. Quinan GV Jr, Kirmani N, Rook A, et al. Cytotoxic T cells in cytomegalovirus infection. N Engl J Med IY82;307:7.
37. Oill P, t:iala M, Schotterman J, ct al. Cytomcgalovirus mononucleosis in a healthy adult. Association with hepatitis, secondary Epstein-Barr virus antibody response and immunosuppression. Am J Med 1977;62:413.
38. Bird AG, McLachlan SM, Birtton S. Cyclosporin A promotes spontaneous outgrowth in vitro of Epstein-Barr virus-induced B cell lines. Nature 1981;289:300.
39. Crawford DH, Sweny P, Edwards J, et al. Long-term T cell-mediated immunity to Epstein-Barr virus in renal allograft recipients receiving cyclosporin A. Lancet 1981;1:10.
40. Magrath IT, Pizzo PA, Novikovs L, et al. Enhancement of Epstein-Barr virus replication in producer cell lines by a combination of 1ow temperature and corticosteroids. Virology 1979;97:477.
41. Abrams D. Lymphoproliferative diseases in homosexual males. In: Purtilo DT, ed. Immune deficiency and cancer: Epstein-Barr virus and Iymphoproliferative malignancies. New York: Plenum press, 1984.
42. Lipscomb H, Tatsumi E, Harada 5, et al. Epstein-Barr virus and chronic lymphadenomegaly in male homosexuals with acquired immunodeficiency syndrome (AIDS). AIDS Res 1 98 3 ; 1:5 9.
43. Birx DL, Redfield RR, Tosato G. Defective regulation of Epstein-Barr virus infection in patients with acquired immunodeficiency syndrome (AIDS) or AlDS-related disorders. N Engl J Med 1986;314:874.
44. Chang RS, Thompson H, Polllerallz S. Epstein-Barr virus in homosexual men with chronic persistent generalized Iymphadenopathy. J Infect Dis 1985;151:459.
45. Sumaya CV, Boswell RN, Ench Y, et al. Enhanced serological and virological findings of Epstein-Barr virus in patients with AIDS and AIDS related complex. J Infect Dis 1985; 154:864.
46. Ragona G, Sirianni MC, Saddu S, et al. Evidence for disregulation in the control of Epstein-Barr virus latency in patients with AIDS related complex. Clin Exp Immuno1 1986;66:17.
47. Rinaldo CR, Kingsley LA, Lyter DW, et al. Association of HTLV-III with EpsteinBarr virus infection and abnormalities of T Iymphocytes in homosexual men. J Infect Dis 19X6; 154:556.
48. Gaston JSH, Richardson AB, Epstein MA. Epstein-Barr virus-specific T-cell memory in renal allograft recipients under long-term immunosuppression. Lancet 1 982; 1 :923.
49. Hanto DW, Sakamoto K, Purtilo DT. The Epstein-Barr virus in the pathogenesis of post-transplant Iymphoproliferative disorders. Surgery 1981;90:204.
50. Groopman JE, Sullivan JL, Mulder (, et al. Pathogenesis of B cell Iymphoma m a patient with AIDS. Blood 1986;67:612.
51. Stahl RE, Friedman-Kien A, Dubin R, et al. Immunologic abnormalities in homosexual men. Am J Med 1982;73:171.
52. Chess Q, et al. Elevation of serum immunoglobulin D (IgD) in patients with the acquired immuno-deficiency syndrome (AIDS). New York Fed Proc 1983;42:6111.
53. Munoz A, Carey V, Saah AJ, et al. Predictors of decline in CD4 Iymphocytes in a cohort of homosexual men infected with human immunodeficiency virus. Journal of Acquired Immune Deficiency Syndromes 1988;1:396.
54. Sonnabend JA, Witkin SS, Purtilo DT. Acquired immune deficiency syndrome (AIDS): an explanation for its occurrence among homosexual men. In: Ma P, Armstrong D., eds. The acquired immune deficiency syndrome and infections of homosexual men. New York: Yorke Medical Books, 1984,409.
55. Stricker RB, Abrams DI, Corash L, et al. Target platelet antigen in homosexual men with immune thrombocytopenia. N Engl J Med 1985;313:1375.
56. Yu JR, Lennette ET, Karpatkin S. Anti-F(ab')2 antibodies in thrombocytopenic patients at risk for acquired immunodeficiency syndrome. J C1in Invest 1986; 77:1756.
57. van der Lelie J, Lange JM, Vos JJ, et al. Autoimmunity against blood cells in human immunodeficiency-virus (HIV) infection. BrJ Haematol 1987;67:109.
58. Andrieu JM, Even P, Venet A. AIDS and related syndromes as a viral-induced autoimmune disease of the immune system: an anti-MHC 11 disorder. Therapeutic implications. AIDS Res 1986;2:163.
59. Ziegler JL, Stites DP. Hypothesis: AIDS IS an autoimmune disease directed at the immune system and triggered by a Iymphotropic retrovirus. Clin Immunol Immunopathol 1986;41:30S.
60. Ashida ER, Scofield VL. Lymphocyte major histocompatibility complex-encoded class II structures may act as sperm receptors. Proc Natl Acad Sci USA 1987; 84:339S.
61. Morris L, Distenfeld A, Amorosi E, et al. Autoimmune thrombocytopenic purpura (ATP) in homosexual men. Ann Intern Med 1982;96:714.
62. Charlesworth JA, Quin JW, MacDonald GJ, et al. Complement, Iymphotoxins and immune complexes in infectious mononucleosis: Serial studies in uncomplicated cases. Clin Exp Immunol 1978;34:241.
63. DeStefano E, Friedman RM, Friedman-Kien AK, et al. Acid labile human leukocyte interferon in homosexual men with Kaposi's sarcoma and lymphadenopathy. J Infect Dis 1982;146:4SI.
64. Buimovici-Klein E, Lange M, Klein RJ, et al. Long-term follow-up of serum-interferon and its acid-stability in a group of homosexual men. AIDS Res 1986;2:99.
6S. Abb J, Kochen M, Deinhardt F. Interferon production in male homosexuals with the acquired immune deficiency syndrome (AIDS) or generalized Iymphadenopathy. Infection 1984;12:240.
66. Buimovici-Klein E, Lange M, Klein RJ, et al. Is presence of interferon predictive for AIDS? (letter). Lancet 1983;2:344.
67. Eyster ME, Goedert JJ, Poon MC, et al. Acid-labile alpha interferon. A possible preclinical marker for the acquired immunodeficiency syndrome in hemophilia. N Engl J Med 1983;309:S83.
68. Preble OT, B1ack RJ, Friedman RM, et al. Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science 1982;216:429.
69. Friedman RM, Preble OT, Black R, et al. Interferon production in patients with systemic lupus erythematosus. Arthritis Rheum 1982;25:802.
70. Hooks JJ, Jordan GW, Cupps T, et al. Multiple interferons in the circulation of patients with systemic lupus erythematosus and vasculitis. Arthritis Rheum 1982.25:396.
71. Skurkovich SV, Eremkina El. The probable role of interferon in allergy. Ann Allergy 1975;35:356.
72. Heremans H, Billiau A, Colombatti A, et al. Interferon treatment of NZB mice: accelerated progression of autoimmune disease. Infect Immun 1978;21:925.
73. Engleman KG, SonnenfieId G, Dauphinee H, et al. Treatment of NZB/NZW Fl hybrid mice with mycobacterium bovis strain BCG or type 11 interferon preparetions accelerates autoimmune disease. Arthritis Rheum 1981;24:1396.
74. Hokland M, Hokland P, Heron I, et al. Se1ective effects of alpha interferon on human T-lymphocyte subsets during mixed 1ymphocyte cultures. Scand J Immunol 1983;17:559.
75. Aune TM, Pierce CW. Activation of a suppressor T-cell pathway by interferon. Proc Natl Acad Sci USA 1982;79:3808.
76. Lindahl-Magnusson P, Leavy P, Gresser I. Interferon inhibits DNA synthesis induced in mouse lymphocyte suspension by phytohemagglutinin or allogeneic cells. Nature New Biol 1972;237:120.
77. Scott GM. Interferons and infectious diseases. In: Taylor-Papadimitriou J, ed. Interferons: Their impact in biology and medicine. New York: Oxford University Press, 1985.
78. Labdevirta J, Maury CP, Teppo AM, et al. Elevated levels of circulating cachectin/ tumor necrosis factor in patients with acquired imn-.unodeficiency syndrome. Am J Med 1988;85:289.
79. Maluish AK, Orta1do JR, Con1on JC, et al. Depression of natural killer cytotoxicity after in vivo administration of recombinant leukocyte interferon. J Immunol t983;131:503.
80. Reddy MM, Chinoy P, Grieco MH. Differential effects of interferon-alpha 2 and inter1eukin-2 on natural kil1er cell activity in patients with acquired immune deftciency syndrome. J Biol Response Mod 1984;3:379.
81. Wu JM, Chiao JW, Maayan S. Diagnostic value of the determination of an interferon induced enzyme activity: decreased 2',5'-oligoadenylate dependent binding protein activity in AIDS patient Iymphocytes. AIDS Res 1986;2:127.
82. Sonnabend JA, Saadoun S, Grierson H, et al. Association of serum interferon with hematologic and immunologic parameters in homosexual men with AIDS and at risk for AIDS in New York City. Abstract 100. Second International Conference on AIDS June 1986. Paris.
83. Grimley PM, Kang YH, Frederick W, et al. Interferon-related leucocyte inclusions in acquired immune deficiency syndrome: loca1ization in T cel1s. Am J Clin Patho1 1984;81:147.
84. Witkin S, Sonnabend JA. Immune responses to spermatozoa in homosexua1 men. Fertil Steril 1983;39:337.
85. Richards JM, Bedford JM, Witkin SS. Rectal insemination modifies immune responses in rabbits. Science 1984;224:390.
86. Mather S, Gaust JM, Wi1liamson HO, et al. Cross reactivity of sperm + T Iymphocyte antigens. AmJ Reproductive Immunol 1981;1:113.
87. Beck BN, Gil1is S, Henney CS. Display of the neutral glycolipid ganglio-N-tetraosylceramide (asialo GM 1) on cells of the natural killer and T lineages. Transplantation 1982;33:118.
88. Witkin SS, Sonnabend JA, Richards JM, et al. Induction of antibody to asialo.GMI by spermatazoa and its occurrence in the sera of homosexual men with the ace quired immune deficiency syndrome (AIDS). Clin Exp Immunol 1983;54:346.
89. Mavligit GM, Talpaz M, Hsia FT, et al. Chronic immune stimulation by sperm alloantigens. Support for the hypothesis that spermatazoa induce immune dysregulation in homosexual males. JAMA 1984;251:237.
90. James K, Hargreave TB. Immunosuppression by seminal plasma and its possible clinical significance. Immunology Today 1984;5:357.
91. McDougal JS, Hubbard M, Nicholson JKA, et al. Immune complexes in the acquired immunodeficiency syndrome (AIDS): relationship to disease manifestation, risk group, and immunologic defect. J Clin Immunol 1985;5:130.
92. Inada Y, Lange M, McKinley G et al. Hematologic correlates and the role of erythrocyte CRl(C3b receptor) in the development of AIDS. AIDS Res 1986;2:235.
93. Tausk FA, McCutchan JA, Spechko P, et al. Altered erythrocyte C3b receptor expression, immune complexes, and complement activation in homosexual men in varying risk groups for acquired immune deficiency syndrome. J Clin Invest 1986;78:977.
94. Jouvin M-H, Rozenbaum W, Russo R, et al. Decreased expression of the C3b/C4b complement receptor (CRI) in AIDS and AlDS-related syndromes correlates with clinical subpopulations of patients with HIV infection. AIDS 1987;l:89.
95. Jensen JR, Jorgensen AS, Thestrup-Pedersen K. Depression of natural killer cell activity by syphilitic serum and immune complexes. Br J Vener Dis 1982;58:298.
96. Sol1ing J, SoI1ing K, Jakobsen KU. Circulating immune complexes in syphilis. Acta Derm Venereo1 (Stockho1m) 1978;58:263.
97. Rook AH, Masur H, Lane HC, et al. Interleukin-2 enhances the depressed natural killer and cytomegalovirus-specific cytotoxic activities of Iymphocytes from patients with the acquired immune deficiency syndrome. J Clin Invest 1983;72:398.
98. Kloster BE, Tomar RH, Spira TJ. Lymphocytotoxic antibodies in the acquired immune deficiency syndrome. Clin Immunol Immunopathol 1984;30:330.
99. Pruzanski W, Jacobs H, I aing LP. I.ymphocytotoxic antibodies against peripheral blood B and T 1ymphocytes in homosexuals with AIDS and ARC. AIDS Res 1983;1:211.
100. Williams RC, Masur H, Spira TJ. I.ymphocyte-reactive antibodies in acquired immune deficiency syndrome. J Clin Immunol 1984;4: 118.
101. Kiprov DD, Anderson RE, Morand P, et al. Antilymphocyte antibodies and seropositivity for retroviruses in groups at high risk for AIDS. N Engl J Med 1985; 312: 1517.
102. SonnabendJA. The etiology of AIDS. AIDS Res 1984;1:1.
103. Sonnabend JA. The acquired immune deficiency syndrome: a discussion of etiologic hypotheses. AIDS Res 1984;1:107.
104. Sonnabend JA, Witkin SS, Purtilo DT. A multifactorial model for the development of AIDS in homosexual men. NY Acad Sci 1984;437:177.
105. Sonnabend JA, Witkin SS, Purtilo DT. The acquired immune deficiency syndrome and Kaposi's sarcoma in homosexual men. In: Cerimele D, ed. Kaposi's sarcoma. Jamaica, NY: Spectrum Publishers, 1985.
106. Sonnabend JA, Witkin SS, Purtilo DT. Acquired immunodeficiency syndrome: Opportunistic infections and malignancies in male homosexuals. JAMA 1983;249:2370.