FACT AND SPECULATION
ABOUT THE CAUSE OF AIDSBy Joseph A. Sonnabend, M.B., B.Ch., M.R.C.P.
AIDS Forum May 1989
INTRODUCTIONThe precise point at which a conjecture comes to be accepted as an established fact is far from clear, although in a commonsense fashion, the distinction between them is usually quite evident. That HIV-1 is the cause of AIDS is a contention that was ceremoniously propelled out of the realm of speculation into that of proven fact by Margaret Heckler in 1984 in her public pronouncement that U.S. government scientists had discovered the "probable cause of AIDS." - HTLV-III, later to be renamed "Human Immunodeficiency Virus," or "HIV," in an apparent confirmation of its etiologic role. Thus, overnight, a new orthodoxy came into being, unruffled by the subsequent discovery that there was a second cause of AIDS in another retrovirus. This required a further adjustment of name; the original virus is now called HIV-1 and the second, HIV-2. HIV-2 is not, as is often claimed, merely another strain of HIV-1, but sufficiently different from it to be considered a distinct although related virus. There are in fact numerous strains of each of these viruses, and this will undoubtedly also be the case for a third HIV, about which a preliminary news report has appeared.
Despite the widespread acceptance of the etiologic roles of the HIVs in AIDS, these must remain conjectural as long as two questions (at least) remain open. One concerns pathogenesis and the other the association of the HIVs with AIDS. Both are important to the original presentation of HIV as the cause of AIDS, as they relate to the two props on which this presentation rested. These were (1) the frequent association of HIV seropositivity with AIDS, and (2) the belief that HIV directly killed helper T lymphocytes, having a demonstrated affinity for these cells, coupled with the acceptance that the depletion of helper T lymphocytes is the hallmark of AIDS. It is now known that insufficient numbers of helper lymphocytes are actively infected to account for their loss by a direct cell-killing effect of HIV, and there is an alternative explanation for the association of HIV seropositivity with AIDS that does not require that it play an etiologic role, and that has yet to be excluded. To these two problems concerning the etiologic roles of the HIVs in AIDS must be added the apparent failure, thus far, of antiretroviral chemotherapy. These issues will be expanded upon in a later section, after a review of some of the far-reaching consequences of inappropriately accepting that the cause of AIDS has been firmly established.
THE COSTS OP CLOSING THE DEBATE ON THE CAUSE OF AIDS
The premature acceptance as fact of a contention that more properly belongs in the realm of speculation has had a number of far reaching consequences-let alone the painful fact that it has provided virtually no help to people with AIDS, despite a massive investment and six years of intensive work on the biology of the HIVs and the chemotherapy of infection with these viruses. Clearly, this is not the only reason for the failure of our scientific and medical institutions to have provided an even rudimentary understanding of the pathogenesis of this disease in the eight years since its first description, let alone to have developed interventionsother than the belated introduction of prophylaxis against pneumocystis pneumoniathat might significantly alter its course. However, it is probably the most important factor as it has justified the almost total commitment of resources to the study of the HIVs and of treatments to counter them.
On the other hand, the study of the HIVs has been spectacularly successful in providing a number of novel insights into the mechanisms of viral replication and, particularly, the control of gene expression. These studies have very significantly advanced the science of virology and cell biology, and will probably find application in the development of methods to control the growth of other viruses.
The acceptance as fact rather than hypothesis that the HIVs cause AIDS is responsible in great part for a number of grave consequences. I will outline some of these in order to illustrate something of the enormous loss that has resulted.
A. The almost total commitment of resources to the study of the HIVs has left alternative etiologic hypotheses unexplored. Should the HIVs be proven not to be the cause of AIDS, we will have to go back to the beginning in our studies on the cause of this disease, and will have lost six years and countless lives.
B. Aspects of pathogenesis apparently unrelated to HIV have not been explored. Some examples will be given that could have been pursued as early as 1981.
1. Cytomegalovirus Infection and Reinfection.
Firstly, the role of infection and reinfection with cytomegalovirus (CMV) a common virus belonging to the herpes group of viruses - in contributing to the immune disregulation of AIDS has been ignored. It was known even before the first description of AIDS that infection with CMV could have several potent immunosuppressive effects. CMV infection is probably universal in homosexual men with AIDS and frequent in other groups of patients. Among the well-documented immunosuppressive effects that can accompany CMV infections are changes in T lymphocyte subsetspredominantly an increase in suppressor cells and a lowering of the ratio of helper to suppressor cells but also sometimes a decrease in helper cells (1,2,3). CMV infections can also be associated with the appearance of circulating immune complexes and of autoantibodies (4,5). Natural killer cell function may be impaired (6) and CMV can also stimulate B cells to over-secrete immunoglobulins (7), and all of these abnormalities are seen in AIDS. However, these defects are attributed to HIV, and the role of CMV in contributing to the immune disregulation of AIDS remains virtually unstudied. CMV has been seen only as an opportunistic invader, playing no part in the underlying immunologic disorder of AIDS. Very recently a report has appeared indicating a more rapid disease progression in hemophiliac patients infected with CMV compared to those who were not (8). Perhaps this will help to belatedly bring attention to the immunosuppressive consequences of CMV infections. Drew had previously shown that CMV infection was associated with a depressed ratio of helper to suppressor lymphocytes in homosexual men (9). To dismiss the possibility that CMV plays a significant role in the pathogenesis of AIDS by asserting that everyone is infected with CMV is not appropriate to conditions where individuals are repeatedly exposed rectally to massive amounts of CMV that can be found in semen. Repeated reinfection with this virus is thus likely, a possibility that has now been shown to occur, at least in gay men with AIDS (10)
2. Epstein-Barr Virus Infection and Reactivation.
A second example of a neglected area of research is the role of reactivated Epstein-Barr virus (EBV) infections in contributing to the immunologic abnormalities of AIDS. Almost all adults will have become infected with EBV, a virus that remains dormant in B Lymphocytes for the life of the individual. However, EBV can be reactivated out of latency, particularly under conditions of depressed cellular immunity, a propensity shared by all viruses of the herpes group, of which EBV is a member. Evidence of frequent EBV reactivations in patients with AIDS and with lymphadenopathy was obtained by our group as early as 1983 (11), but it was years before some limited attention was given to this issue (12,13,14). Whether reactivated EBV infections contribute to the immunologic disorders of AIDS is still a question that remains unexplored. It is in fact quite possible that active EBV infections do contribute to the pathogenesis of AIDS, as such infections are associated with T Lymphocyte subset changes, and can be responsible for increases in suppressor T lymphocytes with a reduction in the ratio of helper to suppressor cells (15). EBV also is a potent activator of B cells, driving them to increased immunoglobulin synthesis (16,17). Thus, EBV may be contributing to the high immunoglobulin levels that are characteristic of AIDS, as well as being associated with the appearance of autoantibodies which are well documented in some EBV infections (18,19,20).
3. The Role of Interferon in AIDS.
Yet another example of a neglected area is the realistic possibility that the sustained presence of high levels of interferon in the circulation of patients with AIDS also contributes to pathogenesis. It has been known since 1981 that people with AIDS have high levels of circulating interferon, a finding we made within a few months of the first description of AIDS (21,22,23). To this day, we do not know what induces interferon in AIDS, nor which cells are making it. It is quite likely that the sustained presence of high interferon levels contributes to the pathogenesis of AIDS, but this possibility remains completely unexplored. Many of the known effects of interferon resemble several features characteristic of AIDS. Interferon can selectively inhibit the proliferation of helper Lymphocytes (24); modulate the activity of B lymphocytes (25) and could contribute to their increased activity; cause elevations in beta2 microglobulin levels (26), a characteristic of AIDS; produce abnormal inclusions in white blood cells, identical to those seen in AIDS (27); activate suppressor Lymphocytes to produce a soluble suppressor of macrophage function (28); cause reductions in white blood cells, red blood cells and platelets (29); regularly cause fevers (29) (in fact, it may be the mediator of the elevated temperature that accompanies virus infections); cause a reduction in trypotophan levels also seen in AIDS (30); and also produce abnormalities in lipids that are characteristically seen in AIDS, namely an increase in serum triglycerides and a lowering of serum cholesterol (31,32).
These are some of the multiple effects of interferon resembling changes characteristically seen in AIDS. Our own studies, reported at the 2nd International Conference on AIDS in Paris in 1986, have shown that high interferon levels are associated with low T helper Lymphocyte counts in AIDS patients (33). Further evidence that interferon may contribute to the pathogenesis of AIDS comes from the observation that low dose naltrexone can lower the levels of interferon and that this lowering is associated with clinical benefit (34). This observation was made by Dr. Bemard Bihari two years ago and has been largely ignored, in all likelihood because it does not directly relate to some hypothetical pathogenic effect of HIV.
Finally, it has been repeatedly noted that the appearance of interferon in the circulation carries an adverse prognostic significance in homosexual and hemophiliac patients (35,36,37).
Perhaps the neglect of the potential role of interferon in contributing to the pathogenesis of AIDS has resulted in part from the desire to administer interferon to people with AIDS as an antiHIV agent, as distinct from its use as an agent against Kaposis sarcoma. Indeed, one of the earliest proponents of using interferon in Kaposis sarcoma now writes "Considerable evidence supports a role for the interferons in both the pathophysiology and therapy of human immunodeficiency (HIV) disease (38)." This statement contains a belated admission that interferon may play a role in pathogenesis and should suggest extreme caution in its use as therapy. If its presence is part of the problem it is not immediately obvious how adding more can be expected to result in improvement. In fact, its administration as an agent against Kaposis sarcoma is without effect in patients who already have interferon in their circulations. It has been claimed that the alpha interferon of AIDS is abnormal and functions differently from conventional alpha interferon. There is absolutely no evidence for functional differences between these interferons. It is now known that the acid lability of the alpha interferon of lupus is conferred by the binding of a protein (39). This is also probably true in AIDS and the identification and origin of this interferon binding protein are other areas awaiting investigation.
4. Autoimmunity.
Yet another neglected area of potentially useful research is the role of autoimmunity. Autoimmunity refers to immune responses directed against the bodys own components.
It was already apparent in 1981 that AIDS had an autoimmune component. We pursued this idea in 1981 following our observation that people with AIDS had high levels of circulating interferon. The reason for looking for autoimmunity was that shortly before 1981, high levels of interferon had been found in the circulation of individuals with lupus and other autoimmune diseases (40,41), and the interferon was of the same type as that seen in AIDS, in having an unusual instability in acid, but, as noted above, with biologic effects identical to the more usual acid stable alpha interferon. Autoantibodies are antibodies directed against self components and are regularly detectable in lupus. Because people with AIDS have the same acid unstable alpha interferon as in autoimmune diseases, we looked for autoantibodies in AIDS and reported their presence in 1983 (42). However, autoimmunity in AIDS has been ignored until only a year or so ago when, in a rather desperate attempt to find a mechanism for HIVinduced pathogenesis, it was proposed that HIV itself is responsible for an autoimmune response which would provide an explanation for the loss of helper T Lymphocytes. This theoretical consideration which has belatedly brought autoimmunity to attention is based on several hypothetical mechanisms. One depends on a similarity between part of HIV and a cell membrane antigen (43). However, a recent review lists many different autoantibodies so far reported in AIDS (44), and it is beyond the capacity of HIV to mimic all the antigens against which these many different autoantibodies are directed.
It should be pointed out that there is really no need to attribute autoimmunity in AIDS to HIV, as it could result from EBV or CMV infections (5,18,19,20) as well as possibly result from immunization by foreign antigens found in blood and semen (45,46). There is indeed evidence that some HIV-seronegative homosexual men have antibodies against T cells (47).
Circulating immune complexes are associations of antibodies and antigens that elicited their production. They are characteristically found in autoimmune diseases, some infectious diseases, including syphilis (48) and CMV infections (49), and are present in AIDS (50,51,52,53,54). Although not proven, they may mediate some of the pathologic changes in AIDS, as they most probably do in the autoimmune diseases. There are several techniques that can reduce the level of circulating immune complexes, just as there are techniques to remove autoantibodies. Some of the few attempts to use these methods in AIDS patients have indeed given encouraging results (54,55,56).
5. Allolmmunization.
Among other unexplored mechanisms that could potentially play a role in contributing to the immunologic disorders of AIDS is the possible effects of alloimmunization. This refers to the immune responses induced by exposure to antigens present on foreign cells in semen and blood. Clearly such exposures could not in themselves produce AIDS, as they are not new; but they are known to produce measurable changes in some immunologic tests and could contribute to disease together with other factors. In fact, multiple blood transfusions are administered to individuals who are to receive a kidney graft in order to make it more likely that their immune systems will not reject the transplanted kidney (57). Among the known immunosuppressive effects of blood transfusions are increases in suppressor T cells, increased suppressor cell activity, and reduced natural killer cell activity (58,59,60,61). In addition, foreign cells can activate latent viruses, including CMV (62) and HIV. In fact, the routine isolation of HIV involves mixing lymphocytes containing dormant and unexpressed HIV with foreign lymphocytes from another individual as a means of activating HIV out of latency. Finally, there is a theoretical possibility that repeated alloimmunizations could lead to the appearance of self-reactive antibodies (autoantibodies).
If active CMV and EBV infections play a role in generating or maintaining some of the immunologic abnormalities of AIDS, even if only to worsen immune function impaired by other mechanisms, we have lost precious time in failing to develop better ways to diagnose active CMV and EBV infections and to develop effective treatments to control these infections. Equally, should interferon prove to contribute to pathogenesis, as is likely, eight years will have been lost in failing to develop methods to control its inappropriate secretion and to remove it from the circulation. Also, the removal of autoantibodies and circulating immune complexes from the circulations of people with AIDS is likely to be of benefit, but there have been only limited, although encouraging, studies of these treatment modalities.
C. A third tragedy stemming from the premature acceptance that HIV has been established to be the cause of AIDS is that virtually all treatment resources have been allocated to the develop. ment of antiretroviral therapiesan approach that has been of precious little help to people with AIDS. IIPA-23 was an effective antiretroviral (63), probably better than AZT, but has helped no one. Suramin (another potent antiretroviral) hurt people, and the benefits of AZT, such as they may be, are transient and could result- from its documented anti-EBV and antibacterial actions. Certainly, by no stretch of the imagination could AZTs benefits be regarded as being of sufficient magnitude to justify such a tremendous allocation of resources to its study.
No treatment models have been pursued other than those based on developing anti-HIV therapies. The use of immunomodulators has been proposed, but the term refers to agents that alter immune functions, either to enhance or depress them. Since so many immune functions are impaired in AIDS, mostly depressed, but in the case of B cells, enhanced, the proposal that immune modulators be tried, without specifying which function is to be modified, and in which direction, is completely Lacking in coherence.
D. Lastly, a terrible consequence of the acceptance as fact that HIV causes AIDS is that patient management strategies that could undoubtedly prolong life have barely been formulated.
People with AIDS die mostly from well recognized opportunistic manifestations. The opportunistic infections can sometimes be prevented, are frequently treatableif not always curableand some can be detected by tests before clinical disease develops. A model for good patient management would have to include measures for the prophylaxis of the opportunistic infections, strategies for the early detection of these infections, as well as for their rapid treatment, the provision of nutritional and psychological support in a structured setting that is optimized for individual patients, and access to help in negotiating the bureaucratic hurdles in obtaining social services and health care benefits. A rapid response to the development of new symptoms requires that patients be well informed and in easy communication with their care providers. The development of patient educational materials to help ensure an early response to an opportunistic manifestation is an appropriate part of the effort to develop patient management strategies.
Virtually nothing has been done to develop such patient management strategies despite the fact that the nature of patient management is probably the most important factor in determining survival - at least in the short term. Prophylaxis for pneumocystis pneumonia had been routinely provided to immunosuppresed individuals even before the onset of the AIDS epidemic (64). This was not regarded as a priority for people with AIDS, and, in fact, it is the community of people with AIDS that must take the credit for bringing about the provision of prophylaxis for the infection that was the most frequent cause of death amongst them. The organized medical and scientific response to AIDS ignored this issue, perhaps in the belief that the development of effective anti-retroviral therapies would make concern for it redundant. Of the other opportunistic infections, cryptococcal meningitis is most probably preventable with fluconazole or itraconazole, cerebral toxoplasmosis with pyremethamine and sulfadiazine, or with pyremethamine alone, tuberculosis with isoniazid, mycobacterium avium intracellulare (MAI) infections with some of the agents used in its treatment, including rifabutin and clofazamine. There is also some evidence that CMV retinitis may be preventable with high-dose acyclovir. It will probably fall to the newly emerging community-based AIDS treatment research organizations to study prophylaxis, as the New York Community Research Initiative (CRI) and the San Francisco County Community Consortium (CCC) performed the studies demonstrating the efficacy and safety of aerosolized pentamidine for the prevention of Pneumocystis pneumonia
The development of improved methods for the rapid diagnosis of opportunistic infections has not received much attention, as is the case for the development of improved methods to treat them.
While not strictly an issue of patient management, a final example of a neglected research area is the treatment of Kaposis sarcoma by agents that can block the angiogenic factors most probably responsible for the growth of this pathological entity. Angiogenesis refers to the new growth of blood vessels, and Kaposis sarcoma lesions contain profile erating but abnormal blood channels. There is evidence that this abnormal Browth is dependent on cell-secreted factors. Purification of these factors and the development of monoclonal antibodies against them is but one approach. The development of drugs to block their action or secretion is another. The search for anti-angiogenesis factors that might suppress the grown of Kaposis sarcoma could have been undertaken years ago, as a reasonable and well known proposal that Kaposis sarcoma is factor dependent appeared in 1975, antedating the onset of the AIDS epidemic (65).
In summary, the acceptance as fact that the HIVs cause AIDS has had the following consequences:
1. Research on other etiologic factors has not been pursued.
2. Aspects of pathogenesis apparently unrelated to HIV have not been investigated. The roles of CMV and EBV infections, alloimmunization, and the sustained exposure to high levels of interferon (and other cytokines, particularly tumor necrosis factor) in contributing to the immune disregulation of AIDS have yet to be explored.
3. Treatment models other than antiretroviral approaches have not been developed.
4. Patient management strategies have yet to be adequately addressed. This issue has been virtually ignored in the belief that an effective antiretroviral would make its consideration redundant.
SOME ADDITIONAL COMMENTS ON WHY THE ASSERTION THAT THE HIVs AS CAUSES OF AIDS MUST REMAIN AN HYPOTHESIS
It has already been mentioned that the etiologic roles of the HIVs in AIDS must remain conjectural as long as at least two issues remain unresolved. The first concerns the possibility that the association of HIV seropositivity with AIDS is without significance regarding the etiologic role of HIV. The second is that proposals concerning indirect mechanisms accounting for HIV-induced loss of helper T lymphocytes remain without support from observations made in vivo. An additional problem is the failure thus far of antiretroviral chemotherapy.
The Association of HIV Seropositivity with AIDS.
There is an explanation for the association of HIV seropositivity with AIDS that does not require that the HIVs play an etiologic role, and that has not been excluded.
Before describing this, it may be helpful to very briefly outline some points about the biology of retroviruses and of the immune response that are relevant. Retroviruses, as is the case with all viruses, consist of a nucleic acid core (in the case of HIV, RNA) surrounded by a protein coat. Retroviruses also possess an outer lipid-containing envelope derived from the outer membrane of the cell in which it was produced. Antibodies are made against the protein components of the virus, not the nucleic acid, although the nucleic acid contains the instructions that can direct the cell to make viral proteins. The amount of protein in a small infecting inoculum may be insufficient to stimulate the body to make antibodies. It is only after the cell has made much more virus that there is sufficient protein to elicit an antibody response. When retroviruses enter a cell, they are disassembled and the nucleic acid is inserted into the genetic material of the cell by a process of reverse transcription in which viral RNA is converted into DNA. This viral DNA may remain completely dormant. In this case, no viral proteins are made and the only indication that the cell contains viral material may be the detection of viral DNA by a variety of techniques, including the newly-developed polymerase chain reaction (PCR) technique.
It had always been dogmatically asserted by AIDS experts that sufficient viral replication follows infection with HIV so that enough viral protein is made to induce an antibody response. Thus, we have been told that after a three month "window" of seronegativity following infection, seroconversion ensues and the infected individual becomes reactive on the HIV antibody test. There is a frequently reproduced graphic representation showing this hypothetical course of events - an initial burst of viral replication after infection followed by the appearance of antibody three months later. However, in the absence of models of human retrovirus infections, there is absolutely no basis to justify this authoritative depiction of the course of infection. It is yet another example of speculation being presented as fact that has typified presentations on AIDS.
It is just as likely that situations exist, perhaps commonly, where infection is followed by very limited viral replication, insufficient to elicit an antibody response, but where the viral DNA is maintained in the cell in a dormant or latent state. In such a case, the individual would be negative on the antibody test, but may show the presence of HIV by a genome detection technique such as PCR If the mechanisms that maintain latency are perturbed (and such perturbation could happen decades after infection, if at all), then the viral DNA is activated, viral proteins are made and assembled into viral particles, and if this process is of sufficient magnitude, the body will respond by making antibodies and thus seroconvert.
The maintenance of latency is probably quite complex and may be perturbed by signals acting on the cell such as interleukin-1 and tumor necrosis factor (66,67), both generated during the course of many different infections. In addition, cells containing latent HIV can be activated to produce virus by contact with alloantigens which are displayed on the surfaces of foreign cells. Also, latent HIV could be activated by superinfecting viruses, particularly herpes viruses (68,69). There also may be immunologic mechanisms whereby cell-mediated immune responses kill cells that start to produce virus, and in this way virus production is limited. The presence of such anti-HIV cell-mediated responses in HIV seronegative individuals should certainly be sought.
If seroconversion depends on the activation of latent viral DNA, then a plausible explanation for the association of HIV seropositivity with AIDS exists that does not require that HIV play any causative role. This is that the expression of the HlVs - viruses that can remain completely dormant in a latent state - represents an opportunistic reactivation resulting from effects, including immune disregulation, that are generated by the true cause or causes of AIDS, whatever these may be, and that these causes are themselves associated with conditions that promote the spread of all microorganisms, pathogenic or not, that can be transmitted between people. The activation of latent microorganisms is indeed one of the characteristics of AIDS.
The determinants of activation (and thus of seroconversion) may be associated with the true cause or causes of AIDS. One proposal is that these causes are to be found, at least in gay men, in an interaction of the effects of repeated CMV infections, reactivated EBV infections and multiple alloimmunizations, as well as other sexually transmitted infections. These exposures could activate HIV by several mechanisms known to do so. Tumor necrosis factor and interleukin-1 are generated during the course of many infections and these substances can activate HIV. (Tumor necrosis factor would also be present in tropical infections particularly malaria and is detectable in the blood of needle-sharing intravenous drug users.) Alloantigens can activate HIV, and gay men, needle-sharing IV drug users and blood transfusion recipients are all exposed to alloantigens. Finally, the immunosuppression associated with CMV, EBV, some tropical infections and alloantigens may impair the cell-mediated immune control of HIV infections, and thereby facilitate HIV production.
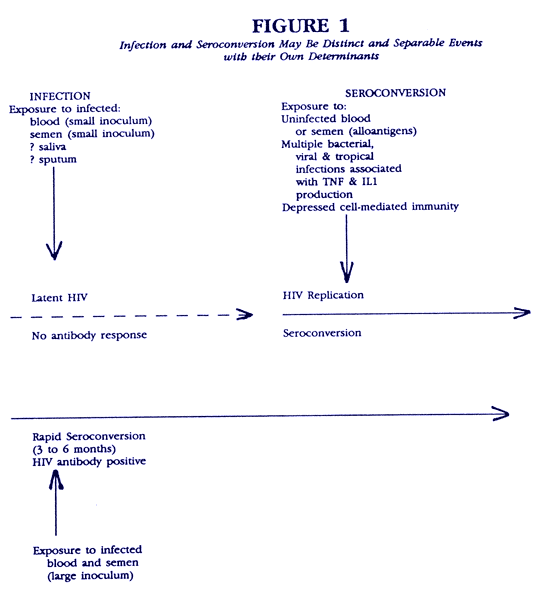
We should therefore separate risk factors for infection with HIV from risk factors for seroconversion. Seroconversion should be thought of as an event separate from infection and with its own determinants. Some conditions may contain risks for both infection and seroconversion, as would be the case, for example, with infections acquired by massive inoculations. Blood transfusions containing large amounts of virus illustrates this possibility. Rectal exposure to semen may constitute a risk for infection if it contains HIV as well as an independent risk for seroconversion in latently infected individuals, even if the semen contains no HIV. There are several potential mechanisms by which seroconversion could be induced by rectal insemination. Firstly, it may expose latently infected cells to alloantigens in the colonic mucosa, or further afield, if cells in semen should enter the blood or lymphatic systems. Secondly, semen may be the vehicle for infection with other viruses, notably CMV, infection with which may trigger activation of latent HIV.
The possible relationships between infection and seroconversion are shown in figure 1.
Thus, conditions which activate HIV may be found in the risk behaviors for AIDS; for example, the acquisition of multiple infections or exposure to multiple foreign antigens, which then are also the risks for seroconversion. As mentioned, the risks for infection with HIV may not be the same as the risks for seroconversion, but although overlapping, extend more broadly. In other words, one may become infected with HIV in ways that are more casual than the contacts required to acquire AIDS (fig. 1). In fact, if HIV is not the cause of AIDS, this might be anticipated. For example, AIDS appears to be spread by an insertive sexual partner and by injection of blood; women appear to transmit AIDS to men with difficulty, if at all. However, it is difficult to comprehend how HIV (as distinct from AIDS) could have been preserred in nature if it were only transmitted by an insertive sexual partner or by blood transfusions. It is thus likely that HIV can be transmitted more easily than AIDS, including its transmission from women to men, and that the virus transmitted without risk behaviors for AIDS will be found in HIV seronegative individuals.
If HIV is not the cause of AIDS, one might expect that groups other than those at risk for AIDS will be found to carry HIV (in the absence of HIV seropositivity). Newer genome detection techniques, such as PCR, may indeed indicate this. Should this be found, for example in health care workers, laboratory personnel working with HIV, or household contacts of AIDS patients, there will undoubtedly be an initial panic that AIDS is indeed spreading out of the groups at Ask; but in fact, such findings must bring further into question the causative roles of the HIVs in this disease.
The association of HIV seropositivity with AIDS could therefore derive from the possibility that the expression of HIV (and consequent seroconversion) is an effect, rather than a cause, of AIDS, and that infection with HIV is more widespread than the distribution of AIDS and of HIV seroprevalence. The above arguments imply that there should be small numbers of people with AIDS who have not been infected with the HIVs.
How Does HIV Cause AIDS?
An important point on which the case for HIV as the cause of AIDS rested was its special affinity for helper T lymphocytes, and the apparently reasonable assumption that this virus was therefore responsible for the depletion of these cells. It was only a matter of time before science would reveal precisely how HIV was killing them. However, it is now known that insufficient numbers of helper T Lymphocytes are actively infected to account for their loss by a direct cell-killing effect of the virus. Therefore, the predilection of HIV for helper T Lymphocytes is of questionable significance with respect to the depletion of this lymphocyte subset.
There is now a desperate search for indirect mechanisms whereby HIV could still, even at a distance, be responsible for the death of helper T lymphocytes. While it is true that a detailed account of pathogenesis is not required in order to attribute an etiologic role to a particular microorganism, proposals concerning possible indirect mechanisms whereby HIV can be responsible for the depletion of helper T Lymphocytes should at least be supported by substantial evidence derived from in vivo observations. None has thus far been presented. For example, a role for giant cells or syncytium formation in accounting for the loss of uninfected cells has been proposed. However, while giant cells may be observed in vitro, they are very rarely observed in tissues of infected patients. A role for free viral envelope glycoprotein in cell-killing linked to an immune response is also proposed, but again no evidence for this can be found in infected individuals. Although there are well-documented precedents for viral-induced autoimmunity, again no evidence exists that HIV can be included amongst them. There are factors other than HIV that could plausibly explain the autoimmune phenomena seen in AIDS.
In conclusion, I have anempted to show why the contention that HIV causes AIDS should be returned to the realm of speculation. The costs of inappropriately accepting that the cause of AIDS has been firmly established to be HIV have been enormous, in time wasted and lives lost. Some of the areas of neglected research have been outlined.
The cause or causes of AIDS remains unknown, and thus all hypotheses, including HIV, must be pursued.
POSTSCRIPT
It has been suggested that questioning the etiologic role of HIV in AIDS may promote the spread of disease as it "frees one of the worry about testing positive or the guilt of spreading the disease"(70). This is an irrational and poorly thought out objection. The reality of the mode of transmission of AIDS, whether sexually or by blood or blood products, is of course quite obvious, whether it is HIV or some other factor or factors that are transmitted. In fact, a ground-breaking booklet presenting the first safer sex guidelines appeared in 1983 (71) and it was based on a multifactorial model (72,73), not a single agent model. The measures suggested were identical to those usually proposed to limit the spread of HIV.
REFERENCES:
1. Reinherz BL, OBrien C et al The cellular basis for viral induced immunodefidency Analysis by monoclonal antibodies. J. Immunol. 1980; 125: 1209.
2. Carney, WP, Rubin RH et al. Analysis of T cell subsets in cyto-megaloviral mononucleosis.J. Immunol 1981; 126: 2114.
3. Verdonck LF, DeGast GC, Is cytomegalovirus infection a major cause of T cell alterions after (autologous) bone-marrow transplantation? Lancet 1984; i: 932.
4. Xantor GL, Goldbey LS, et al. Immunological abnormalities induced by postperfusion cytomegalovirus infection. Ann. Int. Med. 1970; 73: 553.
5. Olding LB, Xingabury DT, et al Pathogenesis of cytomegalovirus infection. Distribution of viral products, immune complexes and autoimmunity during latent murine infection. J. Gen. Virol. 1976; 33: 276.
6. Schrier RD, Rice GP, et al Suppression of natural killer cell activity and T cell proliferation by fresh isolates of human cytomegalovirus. J. Infect. Dis. 1986; 153: 1084.
7. Hun-Fletchet LM, Balachandran N, et al. B-cell activation by cytomegalovirus. J. Exp. Med. 1983; 158: 2171.
8. Webster A, Lee CA, et al Cytomegalovirus infection and progression towards AIDS in haemophiliacs with human immunodeficiency virus infection. Lancet 1989; ii: 63.
9. Drew WL, Mills J, et al. Cytomegalovirus infection and abnormal T-lymphocyte subset ratios in homosexual men. Ann. Int. Med. 1985; 103: 61.
10. Spector SA, Hirata XX, et al. Identification of multiple cytomegalovirus strains in homosexual men with acquired immunodeficiency syndrome. J. Infect. Dis. 1984; 150: 953.
11. Lipscomb H, Tatsumi E, et al. Epstein-Barr virus and chronic lymphadenomegaly in male homosexuals with acquired immunodeficiency syndrome (AIDS) AIDS Res. 1983; 1: 59.
12. Sumaya CV, Boswell RN, et al. Enhanced serological and virological findings of Epstein-Barr virus in patients with AIDS and AIDS related complex. J. Infect. Dis. 1985; 154: 864.
13. Ragona G, Sirianni MC, et al. Evidence for disregulation in the control of Epstein-Barr virus latency in patients with AIDS related complex. Clin. Exp. Immunol. 1986; 66: 17.
14. Rinaldo CR, Kingsley CA, et al. Association of HTLV-III with Epstein Barr virus infection and abnormalities of T lymphocytes in homosexual men. J. Infect. Dis 1986; 154: 556.
15. Crawford DM, Brickell, et al Increased number of cells with suppressor T cell phenotype in the peripheral blood of patients with infectious mononucleosis. Clin. Exp. Immunol 1981; 43: 291.
16. Rosen A, Gergely P, et al. Polyclonal Ig production after Epstein Barr virus infection of human leukoytes in vitro. Nature 1977; 267: 52.
17. Schooley RT, Haynes BP, et al. Mechanisms of Epstein-Barr virus induced human B lymphocyte activation. Cell Immunol. 1980; 56: 518.
18. Pong S, Vaughan JM, et al. Selective induction of autoantibody secretion in human bone marrow by Epstein-Barr virus. J. Immunol. 1982; 129: 1941.
19. Kaplan MB, and Tan BM. Antinuclear antibodies in infectious mononucleosis. Lancet 1968; i: 561.
20. Schooley R. Densen P, et al. Antineutrophil antibodies in infectious mononucleosis. Am J. Med. 1984; 76: 85.
21. Vilcek, J. The importance of having gamma. Systemic lupus erythematosus and Kaposis sarcoma: "Immune" interferon that isnt gamma. In: Interferon 4. 1982, p. 144. 1. Gresser, Ed., Academic Press, Orlando, FL
22. DeStefano E, Priedman RM, et al. Acid labile human leulcocyte interferon in homosexual men with Kaposis sarcoma and lymphadenopathy. J. Infect. Dis. 1982; 146: 451.
23. Preble OT, Rook AM, et al. Role of interferon in AIDS. Ann. N.Y Acad. Sc 1984; 437: 65.
24: Hokland M, Hokland P, et al. Selective effects d alpha interferon on human T-lymphocyte subsets during mixed lymphocyte cultures. Scand. J. Immunol. 1983; 17: 559.
25. Johnson HM. Effect of interferon on antibody formation. Tex. Dep Biol. Med. 1982; 41:411.
26. Pellous M, Bono R, et al. Interferon enhances the amount of membrane-bound beta, microglobulin and its release from human Burkitt cells. Eur. J. Immunol 1981; 11: 524.
27. Grimley PM, Rang YH, et al. Interferon-related leucocyte inclusions in acquired immune deficiency syndrome: localization in T cells. Am. J. Clin. Pathol 1984; 81: 147.
28. Aune TM, and Pierce CW. Activation of a suppressor T-cell pathway by interferon. Proc. Nat. Acad. Sc. USA 1982; 2: 3808.
29. Scott GM. Interferons and infectious diseases. In: Interferons Their impact on biology and medicine. J. Taylor-Papadimitriou, Ed Oxford University Press, New York, 1985.
30. Wemer BR, Puchs D, et al. Tryptophan degradation in patients infected by human immunodeficienq virus. Biol. Chem. Hoppe Seyle 1988; 369: 337.
31. Dixon RM, Borden EC, et al. Decreases in serum high density lipoprotein cholesterol and total cholesterol resuling from natural produced and recombinant DNA derived leucocyte interferons. Metabolism 1984; 33: 400
32. Massaro BR, Borden BC, et al. Effects of recombinant interferon alpha-2 treatment upon lipid concentradons and lipoprotein composidon. J. Interferon Rcs. 1986; 6: 655.
33. Sonnabend JA, Saadoun S, et al. Association of serum interferon with hematologic and immunologic parameters in homosexual men with AIDS and at risk for AIDS in New York City. 2nd International Conference on AIDS. Paris, 1986. Abstract #100.
34. Bibari, B. Low dose naltrexone in the treatment of AIDS. 4th International Conferance on AIDS. Stockholm, 1988. Abstract #3056.
35. Buimovici-Klein E, Lange M, et al. Is presence of interferon predictive for AIDS? Lancet 1983; ii: 344.
36. Byster MB, Goedert J.J, et al. Acid labile interferon. A possible preclinical marker for the acquired immunodeficienq syndrome in hemophilia N. Engl. J. Med. 1983; 309: 583.
37. Metroka CB, Sonnabend JA, et al. Acid labile interferon alpha in homosexual men: A preclinical marker for opportunistic infections. 2nd Intcrnatfonal Conference on AIDS. Paris, 1986. Abstract #101.
38. Rrown, SB. Interferons in human immunodeficiency virus disease: pathophysiologic and therapeutic considerations. Abstract: 1988 Annual Meeting of Tbc International Sociecty for Interferon Researcb. Ryoto, 1988.
39. Yee AM, Buyon JP, et al. Interferon alpha associated with systemic lupus erythmatosus is not intrinsically acid labile. J. Exp. Med 1989; 169: 987.
40. Priedman RM, Preble OT, et al. Interferon production in patients with systemic lupus erythematosus. Arthritis Rheum. 1982; 25: 802.
41. Hooks .IJ, Jordan GW, et al. Multiple interferons in the circulation of patients with systemic lupus erythematosus and vasculitis. Arthritis Rheum 1982; 25: 396.
42. Sonnabend JA, Witkin SS, et al. Acquired immunodefidency syndrome: Opportunistic infections and malignancies in male homosexuals. JAMA 1983; 249: 2370.
43. Golding H, Shearer G, et al. Common epitope in human immunodefidenq virus (HIV) - GP41 - and HLA class II elicits immunosuppressive autoantibodies capable of contributing to immune dysfunction in
HIV-1 infected individualals. J. Clin. Invest. 1989; 83: 1430.44. Kaye BR Rheumatologic manifestation of infection with human immunodefidency virus (HIV). Ann. Inter. Med. 1989; 111: 158.
45. Mather S, Gaust JM, et al. Cross reactivity of sperm and T lymphocyte antigens. Am. J. Reproductive Immunol. 1981; 1: 113.
46. Rubenatein P, Walter M, et al. Immunogenetic findings in patients with epidemic Kaposis sarcoma. In: AlDS andInfections in Homosexual man, p. 403. Ma and Armstrong Eds. Buttenvorth Publishers, Stoneham, MA, 1989.
47. Kiprov D, Andenon RK Antilymphocyte antibodies and seropositivity for retrovirses in groups at high risk for AIDS. NEJM 1985; 312: 1517.
48. Solling J, Solling K, et at al. Circulating immune complexes in syphilis Aeta Derm. Venereol. (Stockholm) 1978; 58: 263.
49. Rundell BB, Bets RP, et al. Interaction of cytomegalovirus immune compexes with host cells. Infect. Immun 1981; 3: 658.
50. McDougal JS, Hubbard M, et al. Immune complexes in the acquired immunodeficieng syndrome (AIDS): relationship to disease manifestation, risk group, and immunologic defect. J. Clin. Immunol. 1985; 5: 130.
51. Buler HH, Kern P, et al. Precipitable immune complexes in healthy homosexual men, acquired immune defidency syndrome and the related lymphadenopathy syndrome. Clin. Exp. Jmmunol 1985; 59: 267.
52. Inada Y, Lange M, et al. Hematologic correlates and the role of erythrocyte CR1 (C3b receptor) in the development of AIDS. AIDS Res 1986; 2: 235.
53. Tausk PA, McCutchan JA, et al. Altered erythocyte C3b receptor expression, immune complexes and complement activation in homosexual men in varying risk groups for acquired immune deficiency syndrome. J. Clin. Invest. 1986; 78: 977.
54. Kiprov DD, Lipper R, et al. The use of plasmapheresis, lymphocytopheresis, and staph protein A immunoadsorption as an immunomodulatory therapy, in patients with AIDS and AlDS-related conditions. J. Clin. Apteresis 1986; 3: 133.
55. Inada Y, Sulzberger J, et al. Removal of ciculating immune complexes by erythrocyte transfusion in patients with AIDS and ARC 4th International Conference on AIDS. Stockholm, 1988. Abstact #2120.
56. Tomar RH, Klorter BB, et al. Plasmapheresis increases T4 lymphocytes in a patient with AIDS. Am. J. Clin. Patbol. 1984; 81: 518.
57. Singal DP, Joreph S, et al. Possible mechanisms of the benefidal effect of pre-transplant blood transfusion on renal allograft survival in man. Transplan. Proc. 1982; 14: 316.
58. Smith MD, Williams JD, et al. The effect of blood transfusion on T suppresaor celh in rend dialysis patients. Transplan. Proc. 1981; 13: 181.
59. Kerman RK, Van Buren CT, et al. Influence of blood transfusion on immune immune responsiveness. Trans. Proc. 1982; 14: 335.
60. Gascon P, Zoumbof, NC, et al. Immunologic abnormalities in patients receiving multiple blood transfusions. Ann. Int. Med. 1984; 100: 173.
61. Pisher P, Lenhard V, et al. Blood transfusion induced suppression of cellular immunity in man. Hum. Immunol. 1980; 3: 187.
62. Olding CB, Jensen FC; et al. Pathogenesis of cytomegalovirus infection I. Activation of virus from bone marrow derived lymphocytes by in vitro allogenic reaction. J. Exp. Med. 1975; 141: 561.
63. Buimovici-Kelin E, Ong KR et al. Reverse transcriptase activity (RTA) in lymphocyte cultures of AIDS patient treated with HPA-23. AIDS Res. 1986; 2: 279.
64. Hughes WT, Kuhn S, et al. Successful chemoprophylaxis for Pneumocystis carinii pneumonitia N. Engl. J. Med 1977; 297: 1419.
65. Warner TPCS, OLoughlin S. Kaposis sarcoma. A byproduct of tumor rejection. Lancet 1975; ii: 687.
66. Osborn L, Kunkel S, et al. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodefidency virus enhancer by activation of the nuclear factor kB. Proc. Natl. Acad. Sc. 1989 86: 2336.
67 Folks TM, Clouse KA, et al. Tumor necrosis factor alpha induces expression of human immunodefidency virus in a chronically infected T cell clone. Proc. Natl. Acad. Sc.. USA 1989; 86: 2365.
68. Albrecht MA, DeLuca NA, et al Herpes simplex virus immediate early protein, ICP4, is required to potentiate replication of human immunodefdency virus in CD4+ lymphocytes. J. Virol. 1989; 63: 1861.
69. Horvat RT, Wood C, et al Transactivation of human immunodeficiency virus promoter by human herpes virus 6. J. Virol. 1989; 63: 970.
70. Gallo, RG HlV-The Cause of AIDS: An overview on its biology, mechanisms of disease induction, and our attempt to control it. J. Acq. Immun. Def. Synd. 1988; 1:521.
71. Callen M and Berkowitz R How to Have Save Sex in an Epidemic: One Approach. Spring 1983, News from the Front Publications, New York.
72. Sonnabend, JA Witkin, SS et al. A multifactorial model for the development of AIDS in homosexual men. Annals N.Y Acad. Sciences 1984; 437:177.
73. Sonnabend, JA, AIDS: An explanation for its occurrence in homosxual men, IN: AIDS and Opportunistic Infections of Homosexual Men, P. Ma and D. Armstrong, Eds., Butterworth Publishers, Stoneham, MA 1989.