Genetica 95: 111-132, 1995
FIVE MYTHS ABOUT AIDS THAT HAVE MISDIRECTED RESEARCH AND TREATMENT
Robert S. RootBernstein
Department of Physiology, Michigan State University, East Lansing, Ml 48824, USA
Abstract
A number of widely repeated and factually incorrect myths have pervaded the AIDS research literature, misdirecting research and treatment. Five of the most outstanding are: 1) that all risk groups develop AIDS at the same rate following HIV infection; 2) that there are no true seroreversions following HIV infection; 3) that antibody is protective against HIV infection; 4) that the only way to treat AIDS effectively is through retroviral therapies; and 5) that since HIV is so highly correlated with AIDS incidence, it must be the sole necessary and sufficient cause of AIDS. A huge body of research, reviewed in this paper, demonstrates the falsity of these myths. 1) The average number of years between HIV infection and AIDS is greater than 20 years for mild hemophiliacs, l 4 years for young severe hemophiliacs, 10 years for old severe hemophiliacs, 10 years for homosexual men, 6 years for transfusion patients of all ages, 2 years for transplant patients, and 6 months for perinatally infected infants. These differences can only be explained in terms of riskgroup associated cofactors. 2) Seroreversions are common. Between 10 and 20 percent of HlVseronegative people in high risk groups have Tcell immunity to HIV, and may haye had one or more verified positive HIV antibody tests in the past. 3) Antibody, far from being protective against HIV, appears to be highly diagnostic of loss of immune regulation of HIV, and some evidence of antibodyenhancement of infection exists. 4) Nonretroviral treatments of HIV infection, including safer sex practices, elimination of drug use, high nutrient diets, and limited reexposure to HIV and its cofactors have proven to be effective means of preventing or delaying onset of AIDS. 5) Many immunosuppressive factors, including drug use, multiple concurrent infections, and exposure to alloantigens, are as highly correlated with AIDS risk groups as HIV. These data are more consistent with AIDS being a multifactorial or synergistic disease than a monofactorial one.
Introduction
Many commentators on AIDS have neatly divided the AIDS world into those who believe that HIV is the sole necessary and sufficient cause of AIDS, and those who believe that it plays no role in AIDS at all. In fact, many investigators believe that HIV definitely plays some role in AIDS, but that its role is as yet undefined. Whether it is necessary but not sufficient to cause AIDS, one of the many factors acting synergistically, or a player in the induction of lymphotoxic autoimmune processes is still an open question (BuimoviciKlein et al., 1988; Donohoe & Falek, 1988; Duesberg, 1987, 1989, 1990, 1992; FernandezCruz et al.. 1988: Haverkos. 1988: Hoff & Peterson. 1989; Hoff et al., 1991; Kion & Hoffmann, 1991; Littlefield, 1992; Lo et al., 1991; Lusso, Lori & Gallo, 1990; Macon er al., 1993; Mathe, 1992; McClean & Nowak, 1992; Montagnier et al., 1990; PapadopulosEleopulos, 1988; Pifer et al., 1987; RootBernstein, 1990a, 1990b, 1990c, 1992a, 1992b, 1993, 1994; RootBernstein & Hobbs, 1993; RootBernstein & DeWitt, 1994; Rubin, 1988, Shearer & Levy, 1984; Sonnabend & Saadoun, 1984; Sonnabend, Witkin & Portillo, 1984; Sonnabend, 1989, Stott, 1991; Wang et al., 1992; Ziegler & Stites, 1986).
The crucial point for any theory of AIDS is to account for not only the presence of HIV and antibodies to it in the vast majority of AIDS patients, but also why some people develop all of the symptoms of AIDS in the absence of HIV, why others remain healthy for decades after HIV infection and may even serorevert, and why the time between HIV infection and fullblown (CDC stage 4) disease varies dously between patients. I will argue that neither the theory that HIV is necessary and sufficient, nor the theory that it is irrelevant to understanding AIDS, is satisfactory for explaining AIDS because both theories are based upon misleading, but widely held myths about the disease and about how biomedical research should best demonstrate the cause(s) of AIDS.
Five myths, in particular, are often repeated in the medical literature, and are demonstrably incorrect: 1) that all risk groups develop AIDS at the same rate following HIV infection; 2) that there are no true seroreversions following HIV infection; 3) that antibody is protective against HIV infection; 4) that the only way to treat AIDS effectively is through retroviral therapies; and 5) that since HIV is so highly correlated with AIDS incidence, it must be the sole necessary and sufficient cause of AIDS. We will not be able to understand and effectively treat AIDS until these myths are exploded and the facts of AIDS installed in their place.
Myth 1: HIV progresses to AIDS at the same rate in all risk groups
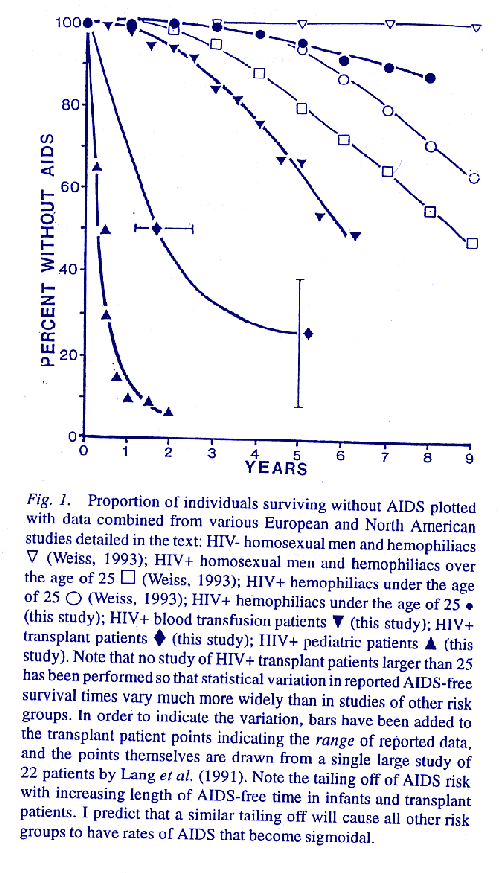
The most powerful and oftencited argument against the need for cofactors and for the sufficiency of HIV is that people in all risk groups progress to AIDS at the same rate following HIV infection. Roy Anderson, for example, states, 'The median incubation period of AIDS (time from infection to the development of AIDS) appears to be approximately 10 years in sexually active adults in developed countries irrespective of risk group' (Anderson, 1993). He, James Curran and many others have argued from such evidence that cofactors associated with particular risk groups cannot have any effect of AIDS progression. Similarly, Robin Weiss states that the development of AIDS is a purely 'stochastic process' and agrees with Anderson 'that population groups with different lifestyles have similar rates of progression to AIDS' (Weiss,1993, his Fig. 2; see my Fig. 1). The one admitted exception are hemophiliacs under the age of 25 who progress more slowly to AIDS than other groups. Weiss claims, however, that after a lag period, they have 'a similar rate of progression to AIDS' as all other risk groups (Weiss, 1993). A review of the relevant literature casts doubt on these crucial claims, however, and thereby on the implication that cofactors are irrelevant to AIDS progression.
Weiss, Anderson, and Curran have evidently failed to examine all of the available studies, or have not paid attention to sophisticated mathematical models of epidemics. For one thing, most hemophilia studies differentiate between hemophiliacs age 2544 and those over the age of 44, because the rate of AIDS development in the over 44 group is two or four times faster than that of the 2544 age group. Weiss can make hemophiliacs have the same rate of AIDS progression as homosexual men only by lumping the 2544 and over 44 age group together. (Weiss, 1993). Furthermore, reference to a large body of studies not cited by Weiss or Anderson reveals a rate of progression to AIDS among hemophiliacs less than 25 years of age that is significantly different from that found in Figure 2 of Weiss's article, and with a slope different than that for homosexual and hemophiliac men in general (Fig. 1) (Derby et al., 1990; Schinaia et al., 1991; Sorice et al., 1989; Giesecke et al., 1988; Eyster et al., 1987).
The discrepancy in the two sets of data is due to reliance by most experts (including Weiss, Anderson, and Curran) on studies performed at hemophilia treatment centers (HTCs) in the United States and Britain. HTCs see only a fraction of all hemophiliacs, generally the most severe cases, but do not see symptomfree hemophiliacs (StehrGreen et al., 1989; Eyster et al., 1987). The majority of the patients studied in the references cited by Weiss (1993), for example, are severe type A hemophiliacs (see Lorenzo et al., 1993 for another example in which 64% of the cohort had severe disease and had exactly the rate of development predicted by Weiss). Less than a third of hemophiliacs have severe disease, however, and the incidence of AIDS in mild hemophiliacs is 1/7 that of severe hemophiliacs (Goedert et al., 1989; Hardy et al., 1985). Thus, incubation periods based on severe hemophiliacs overestimates rate of progression to AIDS, and are not representative of all hemophiliacs (Giesecke et al., 1988). Severity of hemophilia is independently predictive of development of AIDS, possibly due to increased frequency of use of clotting concentrates, transfusions, viral contaminants, and steroidal, colloidal gold, and opiate drugs associated with joint injury treatment (Rosendaal, Smit & Briet, I 991; RootBernstein,1993). In consequence, several studies have shown that reliance on data from HTCs results in large overestimates of rates of AIDS and of total numbers of AIDS cases among hemophiliacs (StehrGreen et al., 1989; Evster et al.. 1987). Overall, only about 15 percent of HIV infected hemophiliacs have developed AIDS in the United States and the United Kingdom at present (CDC, 1993), although one would expect over 40 percent according to the rates of development projected by Anderson and Weiss (Weiss, 1993; StehrGreen et al., 1989).
Further data also contradict the current dogma concerning rate of disease development. Weiss and many other investigators attribute the lower rate of development of AIDS in young hemophiliacs to age. Perhaps, he conjectures 'younger persons have greater CD4 lymphocyte reserves and precursors for renewal' (Weiss, 1993; see also Goedert er al., 1989). Studies of people infected with HIV through transfusions prove this conjecture wrong. Transfusion patients younger than 5 years of age develop AIDS significantly faster than older children, while children over the age of 2 but under 13 years of age develop AIDS at almost exactly the same rate as people between the ages of 13 and 39 and between 40 and 60 (Lui et al., 1986; Medley et al., 1987; Wang & See, 1992; Krasinski, Borkowsky & Holzman, 1989; KopecSchraderetal., 1993).Only people over the age of 59 have a rate of AIDS onset that is shorter than that of younger patients, and the difference is not statistically significant (KopecSchrader et al., 1993). Weiss's conjecture also fails to address why hemophiliacs over the age of 44 should have greatly increased rates of progression to AIDS compared with 2544 year olds when such a phenomenon is not apparent in transfusion patients, nor can it explain why hemophilic men age 20 to 44 years who have additional risk factors such as drug abuse, homosexual activity, or both, have significantly decreased rates of survival (Holman et al., 1992). Moreover, no one has ever reported that age affects the rate of progress to AIDS among HIV infected homosexual men, although a substantial number of these men contract the disease either prior to the age of 25 or after their 44th year. I presume that no age effect has been reported among gay men because no such effect exists although I can find no studies specifically addressing this question.
Those who claim that AIDS develops at the same rate in all risk groups also ignore data showing that transfusion patients of all ages develop AIDS at a significantly greater rate (50 percent have AIDS an average of 5.5 to 7.0 years after infection) (Lui et al., 1986; Medley et al., 1987; Ward et al., 1989; Downs et al., 1991; Msellati et al., 1990; KopecSchrader et al., 1993) than do homosexual men or hemophiliacs (an average of cat 10 years or more). Moreover, the slope of the progression for transfusion patients differs from that seen for homosexual men or hemophiliacs (Figure 1), indicating that the increased rate is not due simply to fewer Tcells at the time of infection, but rather to ongoing synergistic effects with other factors. These factors include the disease process that required surgery in the first place, infection with, or reactivation of, latent immunosuppressive infections including cytomegalovirus, EpsteinBarr virus, and hepatitis viruses, the immunosuppressive effects of anaesthetics, opiate analgesics, chronic and high dose antibiotics, and (following some types of surgery) malnutrition, not to mention chronic health problems that often follow major surgery (reviewed in RootBernstein,1990a, c, 1991, 1992, 1993).
The proof that immunologic status and ongoing exogenous immune suppression are determinants of progression to AIDS comes from studies of organ transplant patients who are infected with HIV (and often other viruses and bacteria) at the time of their operation (Fig. l). Fifty percent of these patients develop AIDS within 1.2 to 2.8 years, depending on the types of transplant and the type of immunosuppressive treatment they receive (Cooper et al., 1993; Schwarz et al., 1993; Ribot&Eslami, 1992;Langeetal.,1991; Tzakis et al., 1990; Atkinson et al., 1987).
It is clear from all of the published studies that the type of immunosuppressive regimen used to control transplant rejection has a very significant effect on the rate at which AIDS develops and on survival, but different investigators have found contradictory results concerning the influence of cyclosporine. Lang et al. (1991) found that four year survival of HIV+ transplant patients treated with the triple drug therapy (azathioprine + steroids + cyclosporine) was 19% compared with 57% in HIV+ patients without cyclosporine. Schwarz et al. (1993), however, have reviewed published cases and claim that the cumulative incidence of AIDS (not survival!) after 5 years was 31 % in patients treated with cyclosporine versus 90% in those receiving immunosuppressi·ve treatments other than cyclosporine. These data may be compatible if one considers the possibility that patients treated with cyclosporine do not generally live long enough to develop AIDS, or that their risk of other forms of death are increased so that AIDS is a less probable diagnosis. Indeed, one problem evaluating all data concerning transplant patients is the very high rate of mortality among these patients regardless of their HIV status. For example, mean survival time for HIV+ dialysis patients, most of whom were intravenous drug abusers in this particular study, was found to be a mere 1.5 years-too short for most of them to develop AIDS (Lang et al., 1991). On the other hand, kidney transplant patients who must return to dialysis because of transplant failure have much better odds of survival than those whose transplanted kidney remains functional (Lang et al., 1991), suggesting that treatments such as plasmapheresis, which have been reported to help some AIDS patients, may indeed be effective. In any event, Lang et al. (1991) summarize their study with words that aptly convey the conclusions of all of the cited transplant studies by saying that 'survival is much lower than in nontransplanted patients contaminated through blood transfusion . .. This shorter survival is due to both a reduced AIDSfree time period and an accelerated course between the diagnosis of AIDS and death.' Clearly, then, chronic treatment with known immunosuppressive agents demonstrably and quite dramatically speeds up the rate of onset of AIDS.
The conclusion that chronic immunosuppressive treatments increase the rate of AIDS development is confirmed by studies of patients who are treated for combined Hodgkin's diseaseHIV infection, and have an average AIDSfree time of only 3 years as a result of cancer chemotherapy (Roithmann et al., 1993). Blood transfusions are also known to be acutely immunosuppressive, and are often given to AIDS patients to correct antiretroviralinduced anaemia. Some evidence suggests that this practice increases the rate at which AIDS progresses (Vamvakas & Kaplan, 1993). One might expect that other immunosuppressive treatments, such as systemic corticosteroids for autoimmune conditions, will also be found to be contraindicated for people at risk for AIDS. On the other hand, nonimmunosuppressivemedical treatments have little effect on AIDS progression, as has been demonstrated by a study of HIVinfected individuals who require cardiopulmonary bypass surgery (Arts, Pomar & Saura, 1993).
Finally, although everyone knows that perinatally infected infants have the fastest rate of progression of any risk group (Weiss, 1993), the huge magnitude of the rate of increase (median of 6 months) (Fig. 1) is often ignored (Blanche et al., 1990; Minkoff et al., 1987; Mayers et al., 1991; Kraskinski, Borkowsky & Holzmann, 1989). This huge increase in rate of AIDS progression has never been explained adequately except in terms of predisposing immunological debilitation caused by maternal cofactors experienced by the fetus or newborn. These factors include drug addiction, multiple concurrent viral and bacterial infections, exposure to blood products and anaesthetics, malnutrition and anemia. A very large proportion of children who develop AIDS are also significantly premature, underweight for their age, jaundiced, and malnourished-all factors associated with immune suppression even in HIVseronegative infants (ibid.; Rubenstein et al., 1983; reviewed in RootBernstein, 1990a, c, 1992, 1993).
I conclude that arguments as to the sufficiency of HIV as the cause of AIDS are not supported by epidemiological evidence. Different risk groups progress to AIDS at substantially different rates, as do different individuals within risk groups. The socalled age factor in hemophiliacs is unique to this group, and must therefore be related to the very significant changes in the treatment of hemophilia over the past few decades (e.g., the switch from heavily virus contaminated plasma, to less contaminated factor concentrate, to ultrapurified and recombinant DNA derived clotting factor), which has saved young hemophiliacs from many cumulative immunosuppressive cofactors that affect older hemophiliacs (Rosendall, Smit & Briet, 1991; RootBernstein, 1990a, 1993). Cofactors known to cause immune suppression and to significantly increase the rates of AIDS development, which are also known to be much more common in older than younger hemophiliacs, include infections (sometimes chronic) with herpes viruses, hepatitis virus types B and C, and cytomegalovirus (Sullivan et al., 1986; Goedert et al., 1989; Webster et al., 1989; Higgins & Goodall, 1991; Sabin et al., 1993) and exposure to alloantigens in impure factor concentrates themselves (Hilgartner et al., 1993; Schulman, 1991; Goedert et al., 1989; Sullivan et al., 1986). All of these cofactors are also present in other AIDS risk groups at unusually high rates compared to the general population (RootBernstein, 1993). Attenuation or elimination of these cofactor exposures through changes in hemophilia treatment since the 1960s resulted in a rise in the average life expectancy of hemophiliacs from 33 years in 1960 to nearly 57 years in 1980 (Aronson, 1988; Rosendaal, Smit & Briet, 1991). (Life expectancy has unfortunately plunged again to 40 years during the period 19871989 due primarily to AIDS, but also to increases in nonAIDSassociated pathologies as well [Lorenzo, et al., 1993; Mares, Sartori & GiroIami, 1992; Ritter, 1994]).
One crucial study that would help to clarify the rateofprogression issue has not yet been done, and that is to examine the rate of AIDS onset in spouses of HIVpositive hemophiliacs and blood transfusion patients as a function both of HIV infection and cofactor acquisition (e.g., exposure to hepatitis viruses, cytomegalovirus, etc.). Presumably, spouses who acquire HIV heterosexually and have no drug or medical problems of their own will have the longest AIDSfree time of any group yet studied. Only two studies have been carried out that are relevant to this issue (Peterman et al., 1988; Andes, Rangan & Wulff, 1989), but neither calculates rate of onset of AIDS, nor gives the elapsed time from HIV exposure. Notably, however, of four women who contracted HIV from their hemophiliac husbands prior to 1985, none had developed AIDS by 1989 and only one had developed AIDSrelated complex in one study (Andes, Rangan & Wulff, 1989), while in the other study, one of 12 spouses had died of AIDS, three had developed lymphadenopathy, and eight had remained asymptomatic by 1988 (Peterman et al., 1988). If one AIDS case out of 16 infections over four to eight years is an approximately accurate interpretation of these results, then the rate of AIDS onset among this group is, indeed, very low.
One interesting observation that has not been made before is that the rate of onset of AIDS in infants and transplant patients appears to level off after a few years, suggesting that those HIVinfected people who remain AIDSfree for extended periods of time have an everdecreasing probability of developing AIDS. The data are not yet conclusive on this point, but if it is true, then one may predict that the probability of developing AIDS in all HIV+ risk groups will come to resemble sigmoidal curves. Longterm HIV+ survivors in all risk groups will, in other words, become less and less likely to develop AIDS. One indication that such a trend has already begun comes from a study of HIV+ gay men in Los Angeles which found that among those infected in 1979, 28% developed AIDS within six years, whereas among those infected in 1983, 25% developed AIDS within six years (Taylor, Kuo & Detels, 1991). The authors of the study suggest three possible explanations: mutation of HIV toward less pathogenic strains; better health care for preAIDS HIVinfected individuals, or control of cofactors through safer sex, clean needle, and education programs. The phenomenon of everslowing progression to AIDS has been observed in homosexual and bisexual men in Amsterdam as well, where the hazard rate has been decreasing consistently (Hendriks et al., 1993).
It is important to stress that cofactor models of AIDS are completely consistent with current data concerning AIDS. In fact, several mathematical models of AIDS epidemiology demonstrate that the observed rates of development in each risk group can be explained only by models involving two or more synergistic agents. Weyer and Eggers (1990), for example, show that even if one assumes the rates of AIDS onset to be the same for each risk group, the 'drastic overrepresentation of the sexually highly active groups and drug abusers in the number of AIDS cases obviously requires that the transmission of AIDS unequivocally depends on the sexual and drug risk ...The observed parallel time series for the spread of AIDS in groups with different risk of infection can be realized by computer simulation, if one assumes that the outbreak of fullblown AIDS only occurs if HIV and a certain infectious coagent (cofactor) are present. Such studies of the accuracy and reliability of Western immunoprecipitation, and polymerase chain reaction (PCR) testing report several cases of seroreversion and loss of detectable virus among groups of tested individuals of varying sizes (e.g., four of twelve cases in Gaffe et al., 1985; four in a hundred in Horsburgh et al., 1990; and one of the 29 in KnuverHopf et al., 1993).
Several very recent reports confirm the existence of unexpectedly large numbers of serorev·erters. Scott Tanenbaum and Cindy Leissinger, researchers at Tulane University found five severe hemophiliacs (representing 10% of their cohort) with HlVseropositive blood samples at one or more time points, all of whom subsequently seroreverted (Tanenbaum et al., 1993; Anonymous, 1994). These researchers point out that between 10% and 15% of severe hemophiliacs who were probably exposed to HIVcontaminated factor one or more times have remained seronegative. Indeed a substantial proportion of people exposed to HIVcontaminated blood factor lots and blood transfusions have also been found to be seronegative on later testing. Henrard et al. (1993), for example, studied 103 high risk seronegative individuals, including 85 hemophiliacs, of whom 52 were known to have been exposed to HIVcontaminated factor concentrates. Seventysix plasma samples (72%) were consistently negative for HIVI DNA by PCR; 24 (22%) were positive only once, but were not so upon multiple retesting; and 4 (3%) were positive twice, and then negative thereafter. When cellular DNAs were extracted from the blood specimens of PRCpositive plasma samples, none were found to be positive for HIV1 DNA, suggesting that the plasmabased PCR test has a very high rate of false positive results when performed on HIVseronegative samples. On the other hand, 10 of 10 PCR tests of cellular DNA from HIVseropositive individuals yielded PCRpositive results. Henrard and his collegues concluded that HIVseronegative individuals who are known to have been exposed to HIV have truly eliminated the retrovirus, and do not remain latently infected.
It is probable that the results reported by Henrard et al. (1993), and Tanenbaum et al. (1993) are very common. Ward and his collegues at the AIDS Program of the Centers for Disease Control studied 765 people who received HIVtainted blood transfusions. Only about 60% were found to be seropositive within an average of 5 years after exposure (Ward e' al., 1989). Similarly, Ludlum et al. (1985) found that of 34 hemophiliacs transfused an average of over a dozen times each with a single batch of HIVcontaminated clotting factor VIII concentrate, only 18 became HIVseropositive, and the risk was not associated with number of transfusions since most of the men seroconverted after receiving only three treatments while others remained seronegative after more than a dozen.
Similarly, Clerici et al. (1992) have documented dozens of instances of seronegativity among high risk individuals, including gay partners of HIVinfected men who have engaged in repeated acts of unprotected receptive anal intercourse, health workers exposed to HIV by subcutaneous cuts and needlestick accidents, and drug users who have shared needles with HIVinfected addicts (Brown, 1992). All of these people, although antibodynegative at the time they were tested, nonetheless displayed active Tcell responses to HIV antigens, suggesting that they had, in fact, been infected previously by HIV. Berkeley researchers have also reported seven healthy people-at least five of whom were definitely exposed to HIV-with Tcell mediated immune responses to HIV, but without HIV antibody. Two of the seven had urine samples positive for HIVantibodies at one time, but seroreverted during the study (Urnovitz et al., 1993).
In addition, many research groups have reported that the existence of significant numbers (hundreds all told) of people who are at least transiently (and sometimes chronically) PCRpositive for HIV, but persistently antibody negative (reviewed in Imagawa & Detels, 1991). These are individuals who have normal immune responses, with normal antibody production. While most of these investigators have interpreted their results as evidence for chronic, latent infection with HIV, Imagawa and Detels have argued that such data are more compatible with the concept of transient or incomplete infection, because subsequent verification of PCR positivity at later time periods is often impossible (ibid.). The transient interpretation is given added weight by the fact that all of the cases reported have been healthy individuals.
Documented cases of seroreversion, in sum, appear to represent the tip of a very large iceberg consisting of thousands of people who have been exposed to HIV and successfully fought off the infection without developing antibodies, or who developed antibodies but seroreverted prior to testing. The reasons for the iceberg being overlooked can be understood if we now go back and reanalyze the methods used by large cohort studies that report extremely low or nonexistent seroreversion rates. These studies all have major flaws in their design. First, most cases of seroreversion seem to occur in people with limited exposure to HIV and who have no ongoing cofactor exposure (FribourgBlanc, 1988; RootBernstein, 1993). People involved in most AIDS cohort studies, on the contrary, are by the design of the studies in high risk groups and very often have continued exposure to both HIV and putative cofactors. Second, the timing of seroreversion appears to be such that many people may have been infected with HIV but seroreverted prior to first being tested for the cohort study, and some appear to remain HIVseronegative during the study despite being exposed to HIV. These people are not identifiable as having been exposed to HIV because of the use of antibody tests for initial screening for infection. Exposure leading to successful Tcell immunity without antibody cannot be documented without the more tedious efforts of Tcell antigen recognition studies, which are very rarely performed. And third, the U.S. Army study is irremediably flawed by the very fact that their data base consists of confirmatory tests, which are normally carried out within a few weeks or months of one another, rather then over the several years that appear to be necessary for loss of HIV antibodies and elimination of HIV from infected cells to occur. Thus, the Armed Forces study is not at all relevant to determining the rate of true seroreversion (Tanenbaum, Leissinger & Garry, 1993).
FribourgBlanc concludes that 'these observations [of seroreversion] are certainly not rare, but authentification of such cases requires conditions that are difficult to satisfy'. His work, and that of an everincreasing number of other investigators tells us that we must stop assuming that if a person is infected with HIV, they will necessarily produce antibody, and that following infection, they will necessarily remain infected forever. For many people, HIV infection is transient, and such people hold the keys to understanding how to combat AIDS.
Myth 3: Antibody to HIV is protective so vaccination is possible
The third myth that I want to discuss is one that is shared alike by those who believe that HIV accounts for all of the immune suppressions in AIDS, and by those (such as Duesberg) who believe that HIV has nothing to do with AIDS. This myth maintains that antibody against HIV is protective, and therefore that the presence of antibody indicates that the retrovirus has adequately been controlled. Duesberg uses this argument in order to contend that since all AIDS patients have high levels of antibody against HIV, therefore HIV cannot be doing any significant damage to the immune system or body (Duesberg, 1989, 1990). Those who believe that HIV is the sole cause of AIDS argue, to the contrary, that if only people can be vaccinated against HIV so that antibodies are present prior to infection, then exposure to HIV will carry no risk of AIDS. Old and new evidence suggests that both of these positions are wrong.
The most important evidence in this regard has already been reported under Myth #2 above, and consists of the fact that HIVseropositivity is usually associated with low CD4 counts that rectify themselves when seroreversion occurs. Additionally, as we have just seen, a significant proportion of people repeatedly exposed to HIV become PCR positive but remain antibody negative and healthy. Other people repeatedly exposed to HIV remain antibody negative, are PCRnegative, but demonstrate Tcell activation toward HIV antigens, strongly suggesting that they have previously mounted an effective Tcell response to HIV (or some crossreactive alloantigen [Stott, 1991; Kion & Hoffman, 1991]). In short, HIV is controlled by the Tcell response, and antibody positivity to HIV is negatively correlated with control of HIV infection.
These data strongly suggest that the primary line of defense, and the only effective one against HIV is a Tcell response (Cleric) et al., 1992; Clerici & Shearer, 1993; Salk et al., 1993). Tcell regulation is reasonably common for noncytolytic encapsulated viruses, and antibody enhancement of infection is relatively common for these viruses as well (see below). Antibody is only produced when Tcell immunity fails to control viral replication and a sufficient amount of free virus is present in blood, lymph, or tissues to activate B cells. Clearly, as the cases of seroreversion prove, this antibody is shortlived in the absence of active, ongoing infection, and does not remain to protect against further infection. This phenomenon of rapid loss of antibody is also consistent with a primarily Tcell regulated immune response, rather than a primarily Bcell regulated immune response.
In fact, there exists a class of infectious diseases, most of which consist of noncytopathic encapsulated viruses (of which HIV is one), the pathological effects of which are actually exacerbated by the presence of antibody. One example is Lymphocytic chor.iomeningitis virus (LCMV) infection in mice, which has many similarities to hepatitis B virus and HIV infection in man (Oehen, Hengartner & Zinkernagel, 1991). In LCMV, as in many other viral infections in which the immune response, rather than the virus infection itself causes lymphocyte death, survival is dependent on a successful Tcell response rather than upon antibody production. Production of antibody, whether naturally occurring or as a result of vaccination, is highly associated with death of the animal (Oehen, Hengartner & Zinkernagel, 1991). Dengue virus infection in human beings presents a similar picture. The severe hemorrhagic fever associated with the virus is almost always a result of an anamnestic or secondary antibody response. Presence of antibody has been found to be a strong predictor of severe disease following reinfection with a variant strain of the virus (Halstead, 1988; Kliks et al., 1989). Since HIV mutates very quickly, and an extremely large number of HIV strains are known, probability of antibodymediated enhancement of secondary infection with variant HIV strains in AIDS becomes a very likely event, which may, indeed, explain the long latency found in the syndrome.
LCMV and dengue virus are only two of many examples antibodyenhanced disease. Porterfield (1986) and Burke (1992) have summarized the relevant data for a very wide range of noncytopathic viruses, including dengue, Japanese encephalitis, yellow fever, tickborne encephalitis, Sindbis, respiratory synctial virus, rabies, reoviruses, murine cytomegalovirus, corona viruses, and lentiviruses (e.g. the visna virus of sheep). In some cases, vaccines (usually live attenuated strains) against these viruses have been very effective, but in others, such as respiratory synctial virus, measles virus, and visna virus, vaccination with formalininactivated whole virus dramatically increased the probability of severe or lifethreatening infection among recipients (Burke, 1992). Burke has concluded that antibodydependent enhancement of infection is a general in vitro property of all enveloped viruses, and that this in vitro activity is more often than not mirrored by physiological enhancement of infection as well, particularly when humoral immunity is not complete.
Particularly frightening in this respect is significant data that antibodydependent enhancement of HIV infection occurs in vitro and possibly in vivo as well (reviewed in Burke, 1992). The problem is exacerbated by the mimicry between HIV proteins and HLA proteins of lymphocytes that results in immunologic crossreactivity between HIV and lymphocyte cellular receptors (Golding et al., 1988; Vega, Guigo & Smith, 1990; Garry et al., 1991; Bjork, 1991; Kion & Hoffman, 1991; Stott, 1991; Clerici et al., 1992; Dalgliesh etal., 1992; SOsal et al., 1993; RootBernstein & Hobbs, 1991, 1993; RootBernstein & DeWitt. 1994).
Thus, a recent study of the immunological effects of recombinant HIV gpl60 resulted in 3 of 5 human volunteers developing antiidiotypic antibodies that cross reacted with their CD4 protein. The study concluded that such vaccineinduced antiCD4 antibodies 'potentially may: (1) limit the use of vaccines which elicit them; (2) contribute to the immunodeficiency occuring in HIV I infected individuals, and (3) provide evidence of HIVI infection during the period when antiHIV1 antibodies are not detectable, (Keay et al., 1992; see also Sabin, 1988).
The presence of nonHIVinduced HIVlike immune responses in several animal models of AIDS (Stott, 1991; Kion & Hoffmann, 1991) also raises the uncomfortable possibility that allogeneic exposure may confound all tests for AIDS, including Tcell tests. Salk et al. (1993) have argued, on the other hand, that allogeneic exposure might be protective, thus providing an alternative explanation for why people with apparent Tcell responses to HIV antigens may have avoided infection (Cleric) & Shearer, 1993; Clerici et al., 1992).
It follows that presence of HIV antibody is symptomatic of a failure of Tcell immunity. The issue in understanding AIDS now becomes that of establishing what causes the failure of Tcell mediated immunity. Since this failure does not occur in a large proportion of people exposed to HIV (e.g., the 15% of severe hemophiliacs who have not become HIV seropositive and perhaps susbstantially higher percentages of mild hemophiliacs), it is unlikely that HIV is, itself, the cause of this failure. HIV is more likely an opportunistic or synergistic infection that becomes manifest only in people predisposed to or with ongoing causes of immune suppression, just as cytomegalovirus and EpsteinBarr virus, which are also extremely highly correlated with AIDS, remain latent in immunologically healthy people, but are reactivated to produce significant viremia and immune suppression in people whose immune systems are suppressed (see references below).
Primary Tcell regulation of HIV explains why HIV is such a good marker for AIDS, regardless of whether it is a causative agent, a synergistic one, or an opportunistic one. If we assume 1) that reexposure to HIV is quite frequent among high risk groups; 2) that exposure to variant HIV strains is therefore common; 3) that only those with concomitant and ongoing immune impairment actually become infected; and 4) that of those infected, only those with ongoing immunological stimulation that adversely effects Tcell control of HIV go on to produce antibody; then HIVseropositivity becomes a very selective criterion for AIDS risk.
One would expect that whatever processes make an active HIV infection possible will also activate other latent viral disease agents. This is, in fact, the case. Both EpsteinBarr virus and cytomegalovirus reactivation and viremia-not the mere presence of antibody-have been reported to be accurate markers of AIDS progression (Bigger et al., 1983; Drew et al., 1985; Fiala et al., 1986; Rinaldo et al., 1986; Rahman et al., 1989; Munoz, et al., 1988; Sumaya et al., 1985), and so have diagnostic signs of autoimmune processes such as the development of lymphocytotoxic antibodies and circulating immune complexes (Cleric) et al., 1992; Zarling et al., 1990; Sonnabend, 1989; Daniel et al., 1989; Ozturk et al, 1987; Stricker, et al., 1987a, b; McDougal et al., 1985). HIV antibody production and viremia, in other words, are only one of a very large set of immunologic events that occur simultaneously in people at high risk for AIDS, and any or all of these events can trigger the very wide range of interferon, interleukin, tumornecrosisfactor, and other immunological events that are often mistakenly attributed solely to HIV (Sonnabend, Wirkin & Portillo, 1984; Sonnabend, 1989; PapadopulosEleopulos, 1988; RootBernstein, 1993; Fauci, 1993).
The treatment implications are manifest. By the time active antibody production against latent viruses has begun in people at risk for AIDS, Tcell disregulation is already severe. Those people who seroconvert immediately following exposure to HIV can therefore reasonably be conjectured to have been previously or concurrently immunosuppressed by nonHIV agents, and I have summarized extremely extensive evidence to this effect elsewhere (RootBernstein, 1990a, c, 1992a, b, 1993). For example, progression to AIDS in hemophiliacs is highly correlated with low Tcell counts (Thelpers < 500) at the time of infection, whereas high Tcell counts (Thelpers >750) are predictive of very slow progression (Lorenzo et al., 1993; Sabin et al., 1993). The object of AIDS prophylaxis must therefore be to prevent the immune system from decaying to the point that seroconversion becomes possible, or to eliminate ongoing factors that prevent seroreversion. It follows that antibodystimulating vaccines may actually be detrimental.
Alternative approaches for treating HIVseropositive people are needed that eliminate ongoing Tcell sup pression and rebuild immunity. Some approaches that embody these principles have apparently been successful, as the next section will outline.
Myth 4: The only way to treat AIDS is to treat HIV
The need for understanding how to treat HIV infections and AIDS raises the fourth myth that has plagued AIDS research. For a decade, the paradigm for preventing AIDS has been to treat HIV, since it is believed that HIV was solely and completely responsible for all aspects of the pathogenesis of AIDS. It is now well established that this HIVcentered paradigm has not yielded any appreciable benefits for AIDS patients and no miraculous antiretroviral drugs or effective HIV vaccines are on the medical horizon (Weiss, 1993; Fauci, 1993). If we accept that AIDS is an immunological disease (rather than a virological disease) in which the major disruption occurs in T cells, and if we accept the fact that different risk groups, and different individuals within risk groups, progress to AIDS at different rates due to different cofactor exposures (see above, Myth #1), and that it is possible for some people spontaneously to eliminate HIV (see above, Myth #2), it becomes manifest that there must be effective approaches to treating people at risk for AIDS that do not depend upon targeting HIV. We need to identify and use these nonHIVcentered approaches to AIDS on the widest possible scale, regardless of whether we believe that HIV is needed to cause AIDS or not, if only because we do not have any effective way of controlling HIV directly at present, and have no real hopes of doing so within the next decade.
One approach that has yielded positive clinical trial results, but little fanfare, is immunologic reconstitution utilizing thymomimetic compounds to treat preAIDS patients (reviewed in Hadden, 1991). Gottlieb et al. (1991) have reported very good results in slowing AIDS progression in a multicenter, doubleblind, placebocontrolled trial of the leukocytederived immunomodulator, IMREG 1, and the pharmaceutical agent has been approved by the U.S. FDA for fullscale clinical trials. Interest has also been increasing in the use of nonspecific potentiators of delayedtype hypersensitivity (DTH) reactions, such as dinitrochlorobenzene (DNCB), that have been found to stimulate Tcell responses, and limited trials suggest some success (Mills, 1986; Stricker, Elswood & Abrams, 1991; Stricker et al., 1993).
A second approach to AIDS is revealed by reviewing of all known cases of true seroreversion (see references in Myth #2 above). Such a review reveals that none of the seroreverters have been treated with antiretroviral drugs. Few. if any, of the cases have been treated for AIDSassociated infections at all. Other factors seem to be operative.
One factor seems to be limited reexposure to HIV. FribourgBlanc (1988) noted that in the cases of spontaneous seroreversion he documented, exposure to HIV was limited to only one or a few instances. Certainly exposure was limited (by the death of the sexual partner) in the case of the hemophiliac wife described by Burger et al. (1985). Similarly, Farzadegan et al. (1988) report that seroreversion in their four gay men followed substantial lifestyle changes that included switching from multiple concurrent and multiple serial sexual partners to a single, stable sexual relationship, and rigorous implementation of safer sexual practices.
Personal interviews with three seroreverters who have provided me with the records of their HIVtesting have revealed that all underwent significant lifestyle changes. All three totally eliminated drug use, began practicing safe sex measures, and went on highnutrient diets. These same factors were reported to be common to all of the longterm AIDS survivors profiled by Michael Callen in his book, Surviving AIDS (1990) and by anecdotal and selfreporting in The Continuum Magazine (England), Praxis (U.S.), and other publications by people with HIV.
Unfortunately, no formal studies of longterm survivors of HIV, or of significantly large groups of seroreverters have yet been carried out, so that it is impossible to say for certain what factors-genetic susceptibility, viral variation, viral load and reexposure, immunological status and at time of infection, subsequent exposure to cofactor infections or. immune impairments from other sources such as chronic medical treatments, nutritional determinants-or other factors are responsible for the difference between healthy seroreversion and fatal development of AIDS. Some clues do exist, however, in a handful of wellcontrolled studies.
Substantial data indicating that elimination of one or more of the nonHIV immunosuppressive risks listed above substantially alters the rate of progression to AIDS in the absence of retroviral treatments. Studies of HIVpositive intravenous drug abusers show that the probability of progression to AIDS can be decreased by a factor of three or more by simply eliminating ongoing drug use (Groenbladh & Gunne, 1989; Weber et al., 1990). Additional treatment for malnutrition and rigorous practice of safe sex procedures decreases the rate of HIV progression among IVDUs even further (Moretti, 1992).
Additional evidence for controllable cofactors in AIDS comes from studies of HIVseronegative hemophiliacs who display pronounced decreases in CD4 counts, decreased capacity to produce interleukin II, and immune suppression (Watson & Ludlum, 1992; Hassett et al., 1993; Madhok et al., 1990; Hay, McEvoy & DugganKeen, 1990). This immune suppression has been related to factor VIII therapy (ibid., and Farrugia, 1992), active cytomegalovirus and hepatitis C infections, and lymphocytotoxic autoantibodies. It is believed that affected hemophiliacs are consequently predisposed to HIV infection and an increased rate of development of AIDS (Sabin et al., 1993; Goldsmitk et al., 1991; Higgins & Goodall, 1991; Madhok et al., 1991; Schulman, 1991; Webster et al., 1989; Daniel et al., 1989). Replacing medium purity blood clotting factor concentrates with high purity, antibody purified, or recombinant factor that lacks viral and alloantigenic contaminates for the treatment of both HIVseronegative and HIVinfected hemophiliacs has resulted in stabilization of Tcell counts, and in some patients, increasing Tcell counts over several years (Hilgartner et al., 1993; Mannucci et al., 1992; Gompert et al., 1992; De Biasi et al., 1991; Schulman, 1991). Increased Tcell counts are a very favorable prognosticator of continued health in HIVinfected hemophiliacs (Daniel et al., 1991).
Another broadly effective approach to AIDS prevention is prophylaxis against cofactor infections. Vaccinations that have proven effective in delaying AIDS onset significantly include Mycobacteria (Kallenius, Hoffner & Svenson, 1989). Prophylactic treatment of cofactor infections such as cytomegalovirus and Pneumocystis pneumonia are also associated with decreased disease progression and also significantly improve survival among HIVseropositive individuals (Odell & Green, 1990; Montaner et al., 1991; Palestine et al., 1991; Hoover et al., 1993). Such results suggest that approaches to AIDS prevention that focus on cofactor prevention, elimination or control may be more effective than treatment of HIV itself and demonstrate that HIV alone is not responsible for AIDS. Thus, development of vaccines against cytomegalovirus, EpsteinBarr virus, hepatitis C, and a set of more effective and less toxic antiviral drugs might be more important for preventing AIDS than an HIV vaccine, and more widely applicable in other medical settings (RootBernstein, 1992b, 1993)
The evidence that cofactors are necessary to the progression of AIDS has now convinced a number of investigators that no comprehensive treatment of AIDS will be possible without addressing the full range of cofactors that may influence disease progression. For example, Lafeuillade and Quilichine (1992) argue that the study of cofactors promises greater 'understanding of AIDS and hence its therapeutic approach'. Littlefield (1992), in an even stronger statement, says that it has now become clear that 'one or more supplemental mechanisms must be involved in the pathogenesis of AIDS,' beyond HIV infection per se, and that 'identification of the nature of this [these] supplemental process[es] has become essential for successful, nonharmful intervention'.
Since data concerning the role of cofactors as regulators of AIDS are limited, four types of tests need to be carried out to confirm the observation that elimination of ongoing immunosuppressive risks (including reexposure to HIV) is effective in preventing progression to AIDS. First, largescale, formal studies of people exposed to HIV without seroconversion, people who have seroreverted, and longterm survivors of both HIV infection and AIDS are desperately needed. Such studies can conclusively demonstrate whether HIV is sufficient to cause AIDS.
Second, a largescale formal study is needed of ongoing immunosuppressive risks among HIVseropositive people progressing to AIDS. It is assumed, but by no means demonstrated, that the immune suppression manifest in such people is due solely to HIV, but no attempt has been made to investigate ongoing immunosuppressive exposures.
Third, a study of people exposed to the same sorts of immunosuppressive agents in the absence of HIV is mandatory. It is well established that all risk groups have some degree of immune suppression independent of exposure to HIV (reviewed in RootBernstein, 1993). No study has ever been carried out that has examined HIVpositive and HIVnegative individuals in the same risk groups, paired by ongoing nonHIV immunosuppressive risks. Do all severe hemophiliacs actively infected with hepatitis B virus, cytomegalovirus, EpsteinBarr virus, and using intermediate purity clotting factor experience the same Tcell depletion regardless of HIV infection, or not? To what degree is their Tcell depletion greater than or less than uninfected hemophiliacs, or those who use ultrapure factor, regardless of HIV status? (That we should even have to ask for such studies a decade after the discovery of HIV is a symptom of how poorly controlled and designed research on AIDS has been).
Fourth, a converse study is also necessary that could potentially prove that HIV is both necessary and sufficient to cause AIDS: I have challenged the medical community for four years now to find me a significant number of people (say 100 or more) with AIDS who have HIV as their sole immunosuppressive risk (as defined by the set of clinically recognized factors known to cause immune suppression that are listed in my book, Rethinking AIDS [RootBernstein, 1993]), whose Tcell counts and other immunological parameters were normal at the time of HIV infection, and who have nonetheless developed Tcell counts below 500 and an opportunistic infection. So far, no one has responded to this challenge. Surely it is not too much to ask that a hundred such cases be found among the hundreds of thousands of AIDS cases that have been documented, if we are to accept the oftrepeated assertion that HIV alone is sufficient to cause AIDS.
One caveat that must be understood in setting up the abovementioned studies is that AIDS itself (as opposed to HIV infection) may not be reversible. AIDS may involve slowly progressive processes including vicious cycles in which multiple sets of infections coactivate one another, and autoimmune processes that inexorably cause immune system or other forms of systemic decay. It may not be possible to reverse AIDS by simple remediation of risk factors after a certain point in the disease, as both Sonnabend and I have pointed out.
We will need, in addition to preventative measures against AIDS, new, nonretroviral approaches to disease treatment of multiple, concurrent infections (many of which are not susceptible to individual antibiotics), ways to reverse autoimmune processes, and methods to reconstitute immune function. The frightening fact is that very little of this very necessary research has even begun. The HIV paradigm has blinded most researchers to the undeniable fact that every AIDS patient (as opposed to HIVinfected person) has one or more autoimmune diseases, and we do not know how to treat adequately, let alone cure, any human autoimmune disease; and that every AIDS patient, even if cured of their HIV infection, would still have sustained possibly irreversible immunologic damage. It is entirely possible that we may one day learn how to vaccinate against or eliminate HIV in AIDS patients and still have people dying of AIDSassociated autoimmune conditions and opportunistic infections simply because we have nor yet begun to investigate the causes or cures of the relevant immuno1ogic processes (RootBernstein, 1992a, b, 1993; Root-Bernstein & Hobbs, 1993).
Myth 5: AIDS must have a single etiologic agent
Another myth that has adversely affected AIDS research is the assumption, common to much of modern biomedical research, that every disease has a single etiological agent. As powerful as the onegerm/onedisease/onecure paradigm has been in the development of modern medicine, it has now been admitted by most HIV experts that the immune system destruction observed in AIDS cannot be accounted for by any known mechanism of direct HIV pathogenicity (Cohen, 1993; Fauci, 1993; Gougeon & Montagnier, 1993; Littlefield, 1992; Lo et al., 1991). Surprisingly, this admission has not, however, caused any major AIDS researchers to question whether HIV is therefore sufficient to cause AIDS. There are, however, other paradigms for disease etiology that are relevant to AIDS besides the onegerm paradigm that are fully consistent with existing data, as Sonnabend and Saadoun pointed out as early as 1984 (Sonnabend & Saadoun, 1984). Unfortunately, these alternatives are less wellknown than the onegerm/onedisease dogma and their implications have been generally ignored (RootBernstein, 1993).
The least revolutionary alternative to the onegerm paradigm is the twogerm or infectious synergism paradigm (reviewed in RootBernstein, 1993). Perhaps the earliest proven case of virusbacterium synergism was discovered by Shope in 1931, when he showed that neither Hemophilus influenzae nor an unidencified virus, each of which could be isolated from 100% of pigs who contracted fatal swine flu, was capable of causing disease in healthy animals. A combination of the virus and bacterium, however, nearly always caused a fatal pneumonia (Shope, 1931). A similar synergism was found by Dudding et al. (1972) between adenovirus and several bacteria, including Hemophilus influenzae, which caused fatal pneumonias in man. Huang and Hong (1973; and Huang et al., 1973) found that multiple viral infections often resulted in the production of lymphocytotoxic antibodies in human patients, accompanied by significant immune suppression. Hamilton, Overall and Glasgow (1976) found similarly that combinations of murine cytomegalovirus with Pseudomonas, Staphylococcus, or Candida infections were much more likely to be fatal to mice than individual infections, even when the combined doses of infectious agents were thousands of times smaller. Another wellestablished human example is the combination of influenza virus with Staphylococcus aureus to yield a severe and often lethal pneumonia (Klenk & Rott, 1988). It is likely that the terrible influenza epidemic that killed so many people after World War I was in fact the result of two or more epidemics (one influenza, the other(s) unidentified bacteria that overlapped.
Significant evidence exists to suggest that HIV may interact synergistically by mechanisms such as transactivation or immunologic crossreactivity with other infectious agents associated with AIDS, including herpes simplex viruses, cytomegalovirus, EpsteinBarr virus, HTLVI, mycoplasmas, and mycobacteria (Littlefield, 1992; Lo et al.. 1990, 1991; Lussoetal., 1990, 1991; Montagnier et al., 1990; RootBernstein, 1990a, 1992a, 1992b, 1993; Wang et al., 1992). All of these infectious agents are present in people in AIDS risk groups and in people with AIDS at incidences hundreds of times higher than in the general population (BuimoviciKlein et al., 1988; Macon et al., 1993; RootBernstein, 1993; Sonnabend, Witkin & Portillo, 1984; Sonnabend, 1989).
A second alternative to the onegerm model is the multifactorial or predisposition model. HIV may be an opportunistic agent that, like Pneumocystis carinii or cytomegalovirus, is not deadly except in immunosuppressed individuals. In this case, a broad set of immunosuppressive agents, including many addictive drugs, malnutrition, immunosuppressive infectious agents, and exposure to alloantigens in semen, blood, blood products, or contaminated needles may interact to set the stage for active HIV infection (Sonnabend, Witkin & Portillo, 1984; Sonnabend, 1989; Donohoe & Falek, 1988; Duesberg, 1990; FernandezCruz et al., 1988; PapadopulosEleopulos, 1988; Pifer et al., 1987; RootBernstein, 1990a, c, 1993).
A third alternative model is an autoimmune model for AIDS. Virtually all people with AIDS have lymphocytotoxic antibodies present. Such antibodies are associated with a number of agents, including cytomegalovirus infection, exposure to blood and blood products, and immunologic exposure to semen, in both HIV and nonHIV infected individuals (Huang & Hong, 1973; Huang et al., 1973; reviewed in RootBernstein, 1990b, 1992 a, b, 1993; RootBernstein & Hobbs, 1993; RootBernstein & DeWitt, 1994; RootBernstein & DeWitt, this volume). Significant debate surrounds the issue of whether HIV is necessary or sufficient to induce lymphocytotoxic autoimmune processes in AIDS, but significant data point to alloantigen and infectious agents other than HIV as targets for lymphocytotoxic antibodies in AIDS patients, in addition to HIV (Bjork, 1991; Dalgliesh et al., 1992; Garry et al., 1991; Kion & Hoffman, 1991; Stott, 1991; Ziegler & Stites, 1986; Sonnabend, 1989; Hoff & Peterson, 1989; Hoff et al., 1991; Zarling et al., 1990; Morrow et al., 1991). I, personally, believe that the only way to explain any autoimmune disease, including the type that appears in AIDS, is by means of mutipleantigenmediated induction (RootBernstein, 1990b, 1991, 1993; RootBernstein & Hobbs, 1991; 1993).
It is extremely important to realize the statistical implications of multipleagentinduced diseases (MAIDs), whether they are cofactorial, synergistic or autoimmune, for the epidemiology of such diseases. Very simply, MAIDs grow at rates that are multiplicative functions of the rate of growth of the individual agents. If, for example, two synergistic disease agents are each present in 1 in 1000 people, and the diseases are randomly distributed, then the probability that they will coinfect an individual is 1 in a million. Increasing the incidence of each agent by a factor of ten (so that each is now present in 1 in 100 people) results in a probability of combined infection of 1 in 10,000. In other words, a 10fold increase in the incidence of individual disease agents results in a 100foldincreased probability of acquiring a combined infection. Increasing the incidence of each agent by a factor of 100 (so that 1 in 10 people are infected) results in a probability of combined infection of 1 in 100, a 10,000fold increased probability of coinfection. Thus, relatively small increases in the incidence of cooperative agents lead to extremely large increases in the incidence of the MAID they cooperatively cause. If the two diseases are cotransmitted, or are transmitted in a nonrandom way such as occurs in high risk groups for AIDS, then the probability of coinfection is obviously increased even further (RootBernstein & Hobbs, 1993). The MAID theory may therefore explain the observation that people in the advanced stages of AIDS seem to be more of an infectious risk to their partners than are people with uncomplicated HIV infections (European Study Group, 1989; Padian, Shibosla & Jewell, 1990): people with fullblown AIDS are, by definition, multiply infected.
The application of the MAID concept to AIDS is illuminating. One of the most important questions concerning the current epidemic of AIDS is whether HIV is an old or new virus. A variety of evidence suggests that both HIV and AIDS have existed in human populations for decades, and probably for centuries, prior to the recognition of the first AIDS cases or the discovery of the virus (reviewed in RootBernstein, 1990a, c, 1993). Why, then, did AIDS become epidemic only during the 1980s? One possibility, for which extensive evidence exists, is that the modes of transmission grew exponentially during the previous decade as can be verified by huge increases in all sexually transmitted diseases and drug use (both intravenous and otherwise) (Duesberg,1992; RootBernstein, 1993). But these increased modes of transmission spread not only HIV, but many of its putative cofactors-whether they consist of coinfections, drugs, or exposure to alloantigens or all three-simultaneously. The simultaneous spread of HIV and cofactors (consider the cotransmission of hepatitis viruses and HIV in gay men during the late 1970s), in this case nonrandomly in specific risk populations, would have resulted in increases several orders of magnitude larger in the incidence of any disease manifestations that are multipleagent dependent. (Recall, on this point, that the incidence of HIV seropositive individuals must represent only a fraction of the people actually exposed to HIV-the fraction that did not successfully combat HIV without developing an antibody response, or who subsequently seroreverted-see Myth #2 above. Persistently HIV seropositive individuals therefore probably represent people who have ongoing cofactor exposure). I have, in fact, demonstrated that the incidence not only of HIV, but of active cytomegalovirus and EpsteinBarr virus infection, hepatitis viruses, Mycoplasma infections, addictive drug use, lymphocytotoxic antibodies, and various other immunosuppressive agents is often hundreds of times higher among high risk group populations than among low or nonrisk heterosexuals and lesbians (RootBernstein, 1993), which would make the incidence of MAIDs tens of thousands of times higher among risk groups then in the general population. Thus, if AIDS is a MAID, both the time course of the epidemic, its exponential growth, and its maintenance within specific high risk populations can be explained by the unusual coincidences of both HIV and all of its possible cofactors. If, on the other hand, AIDS is caused solely and simply by HIV, then neither the timing of the current epidemic nor its specificity for risk groups is comprehensible.
It should also be noted that a direct consequence of considering etiologies for AIDS that are more complex than a single agent immediately invalidates two standard approaches to determining disease causation: epidemiological correlations and Koch's postulates. If AIDS is more complex than a simple HIV infection, then the high correlation between HIV and AIDS (Koch's first postulate) is not sufficient to demonstrate that HIV is the causative agent. The observed correlation strongly suggests that HIV is one part of the cause of AIDS, but it should be expected that other immunosuppressive agents and/or behaviours will be as highly correlated with either all AIDS cases, or with individual risk groups. Thus, it is not surprising to find that statistically significant correlations exist between AIDS risk and unprotected receptive anal intercourse in both men and women (Naz et al., 1990; Adams et al., 1988; Morrow et al., 1991; Sonnabend, 1989; reviewed in RootBernstein & DeWitt, this volume); to use of inhalant nitrites by gay men (Vandenbroucke & Pardoel, 1989; Haverkos, 1988); to general use of drugs in all risk groups (Duesberg, 1992); and to the incidence of active infection (not presence of antibody) with cytomegalovirus, EpsteinBarr virus, and mycoplasmas in all risk groups (BuimoviciKlein et al., 1988; Munoz et al., 1988; Rahman et al., 1989; Rinaldo et al., 1992; Lo et al., 1992; Wang et al., 1992). But just as HIV is not sufficient to cause AIDS, so, apparently, are these agents not sufficient to cause AIDS. Thus, demonstration that people with exposure to these nonHIV agents do not get AIDS in the absence of HIV does not prove that they are not part of the cause of AIDS. What must be tested, both epidemiologically and experimentally, are their combinations. Moreover, there is no requirement in a multifactorial disease that one specific pair of agents be necessary to cause the disease. One agent may have several different cofactors, none of which correlate highly with the disease. Or there may be several combinations of agents that can create the same pathological symptoms (as, for example, in the case of pneumonia), and therefore no single agent or set of agents that correlates one hundred percent with all clinically diagnosed cases of the disease.
What is at issue here are the criteria for evaluating disease causation. Koch's postulates, focusing as they do on a single etiologic agent, do not apply to diseases that are caused by multiple, interacting agents. Thus, the reason why HIV has failed to satisfy Koch's postulates, despite its obvious presence in virtually all AIDS cases, may have nothing to do with the oftcited species specificity of HIV (an observation belied by the fact that HIV does infect and replicate in chimpanzee and macaque lymphocytes) but rather may result from the fact that Koch's postulates are irrelevant to testing AIDS etiology (RootBernstein, 1992b, 1993). HIV may not be able to cause AIDS because it is not sufficient to cause AIDS. This is no different than saying that neither influenza virus or Staphylococcus are sufficient to cause pneumonia. But both are necessary.
Diseases such as AIDS that are autoimmune in nature, or are the result of synergistic or multifactorial processes require the satisfaction of a set of postulates other than Koch's (Witebsky et al., 1957; RootBernstein, 1991). For example, MAIDs may be characterized by satisfying the following criteria: '(1) two or more infections or immunosuppressive agents will be active simultaneously in individuals affected with a particular disease; (2) people with only one of these agents will either have no symptoms of the disease or symptoms of another disease that is associated with the single agent; (3) these multiple agents will be isolatable or identifiable individually; (4) no single one of these agents will be capable of inducing the disease by itself when introduced into healthy human beings or animals (although they cause different disease symptoms); (5) an immune response to each agent will be demonstrable during disease induction and will be significantly altered by the combination of agents as compared with any agent individually; (6) the disease will be transmissible from one animal or human being to another by means of an appropriate combination of the putative causative agents. In the case of autoimmune diseases, the transmissible factors may include tissue or purified proteins, immune serum, lymphocytes, other cells or antibodies' (RootBernstein, 1993). As far as I am aware, no one has yet set up an animal model of AIDS that would satisfy these criteria, and therefore model AIDS as a MAID. We cannot, therefore, rule out the possibility that AIDS is caused by some combination of agents, of which HIV is likely to be one.
The fact is that until someone is able to produce an animal model of AIDS using the exact set (or sets) of agents found in human beings (not merely analogous agents such as feline or simian immunodeficiency virus, for example), we will not know whether we are working with correct or incorrect theories of causation. My tendency is to believe that having failed to cause AIDS with HIV alone, we must assume that AIDS is more complicated than a mere retroviral infection. On the other hand, the same reasoning makes me reject Duesberg's suggestion that opiate or other drug abuse is the cause of AIDS: investigators have worked with animal models of addiction for decades without observing AIDS in their animals. Thus, AIDS is more than just HIV or just drugs or just any single agent that we currently understand. We must now build on what we know of HIV, but in such a way as to expand our range of research to look at how it behaves in the much more complicated conditions that actually exist in real AIDS patients with multiple infections, blood product exposures, drug exposures, malnutrition, and alloantigen exposure. These conditions have never been duplicated in animals or test tubes.
On this final point I must insist upon the accuracy of the following observations. First, there is no documented case of anyone who has developed AIDS who does not have several of the following immunosuppressive agents at work prior to, concomitant with, or following their HIV infection: multiple, concurrent infections with identified immunosuppressive viruses and bacteria (e.g. herpes viruses, hepatitis viruses, and mycoplasmas); immunologic exposure to alloantigens (e.g., semen, blood or lymphocytes); chronic or high dose treatments with antibiotics; anaesthetics; chronic or high dose use of immunosuppressive addictive drugs irrespective of mode of use (e.g., heroin); malnutrition; and autoimmunity directed at Tcell subsets (reviewed in RootBernstein, 1990a, c; 1992 a, b; 1993). As a result of these immunosuppressive risks, many hemophiliacs, blood transfusion patients, drug abusers, infants of people in these risk groups, and homosexual men are significantly immunosuppressed even in the absence of HIV infection (reviewed in Duesberg, 1992; RootBernstein, 1990a, 1993). Second, and conversely, there is no evidence that HIV can cause disease in an immunologically healthy person free of these causes of immune suppression: people with limited exposure to HIV and no ongoing risks of the sort just enumerated, do not become infected with HIV or serorevert (see above); and there appear to be no verifiable tertiary cases of AIDS (i.e., nonrisk group heterosexual to heterosexual transmission) in Western nations. AIDS is therefore remaining in identified risk groups (National Research Council, 1993; Fumento, 1990)-a fact that cannot be explained except if prior immune suppression or cofactors are necessary for HIV transmission and seroconversion.
New directions in AIDS research
The recognition that AIDS may be more complex than HIV is important because it allows us to reevaluate existing data in new ways that reveal both new problems and new solutions to the epidemic. For example, any multipleagentinduced disease (MAID) can be eliminated by controlling any of the multiplicity of necessary agents. Thus, we need not target only HIV in order to control AIDS, if AIDS can be demonstrated to be a MAID. Both HIV and its cofactors (see Myth #4 above) become viable targets for prophylaxis. treatment and cure. Indeed, one of the reasons that current public health policies such as safer sex, elimination of drug use, and clean needle exchanges may be working effectively in many communities (Van Griensven et al., 1989; Judson, 1990; Winkelstein et al., 1988; Weber et al., 1990) is that both HIV and many of its cofactors are simultaneous!) being controlled. According to the MAID theory, the result of simultaneous control should be a reduction in new AIDS cases that is a multiplicative function of HIV and cofactor prevalences. This possibility mandates a much higher interest in nonHIV factors associated with AIDS than has thus far been manifest.
Beyond the obvious point that there are models of disease causation (and therefore prevention and treatment) that have been ignored by the majority of AIDS researchers lies an even more important point: scientific research consists of elaborating all of the possible explanations of a phenomenon and then eliminating, through controlled observation and experiment, all but one of these (RootBernstein, 1989). Because this is the way all good science works, philosophers and practitioners of science both agree that one can never prove that the remaining answer is true; one can only prove that all of the other possibilities are not (Popper, 1962; Medawar, 1967). (Anyone who remembers learning Mendelian genetics will recall that the key to solving genetics problems is not determining what type of inheritance explains any particular cross, but remembering all of the different possibilities and searching for the crucial evidence that makes all but one of them untenable). Good scientific research therefore consists primarily of performing experiments that disprove theories', rather than gathering data that purport to support a preconceived notion.
In pointing out that too much AIDS research has been directed by myths that have been accepted uncritically by the majority of investigators, and that several wellestablished models of disease causation have been ignored as well, I am therefore arguing that AIDS research as a whole has not followed standard scientific practice of skeptical elaboration of possibilities followed by disproof. Instead, the community of AIDS researchers has reversed standard practice by leaping to the conclusion that the first obvious answer (HIV) must also be the best and the most correct answer. Until it can be demonstrated that no other possible explanation of AIDS exists besides HIV alone, and until specific tests are performed to attempt to disprove the HIV theory, there is no methodological justification for limiting research to HIV alone. This is basic scientific method. Einstein had some advice to scientists. He said that when devising a theory, make it as simple as possible and no simpler. My view of AIDS research, which owes a great the pioneering work of Joseph Sonnabend, is that both those who study HIV and those who refuse to acknowledge its role in AIDS have oversimplified. AIDS is complex. AIDS is multifactorial, and the diverse factors that are correlated with the various risk groups for AIDS all interact synergistically. Until we understand these interactions in their full complexity and set up appropriate experiments to test whether any of them are relevant to AIDS pathogenesis, we will continue to act, as we act today, like the blind men describing the elephant, each attributing all of AIDS to the part of the thing with which we are in closest contact (RootBernstein, 1993). Meanwhile, people with AIDS die as a result of our blindness.
References
Adams, L.E., R. DonovanBrand, A. FriedmanKien, K. Ramahi & E.V. Hess, 1988. Sperm and seminal plasma antibodies in acquired immune deficiency (AIDS) and other associated syndromes. Clin. Immuno. Immunopathol.46: 442449.
Anderson, R., 1993. AIDS: trends, predictions, controversy. Nature 363: 393394.
Andes, W.A.. S.R. Rangan & K.M. Wulff, 1989. Exposure of heterosexuals to human immunodeficiency virus and viremia: evidence for continuing risks in spouses of hemophiliacs. Sex Transm. Dis. 16: 6873.
Anonymous, 1994. Fighting HIV: A clue from hemophilia. Science 263:27.
Aris, A., J.L. Pomar & E. Saura, 1993. Cardiopulmonary bypass in HlVpositive patients. Ann. Thorac. Surg.55: 11041107.
Aronson, D.L., 1988. Cause of death in hemophilia patients in the United States from 19681979. Am. J. Haematol. 27: 712.
Atkinson, K., A.J. Dodds, All. Concannon & J.C. Biggs, 1987. The development of the acquired immunodeficiency syndrome after bone marrowtransplantation. Med. J. Aust. 147: 510512.
Bames, D.M., 1988. Obstacles to an AIDS vaccine. Science 240: 719721.
Biggar, R.J., H.K. Anderson, R Ebbesen, M. Mebye & J.J. Goedert, 1983. Seminal fluid excretion of cytomegalovirus related to immunosuppression in homosexual men. Br. Med. J. 286: 20102012.
Bjork, R.L. Jr., 1991. HIVI: seven facets of functional molecular mimicry. Immunology Lett. 28: 91 95.
Blanch, S.. M. Tardieu, A. Duliege. C. Rouzioux, E le Deist, et al., 1990. Longitudinal study of 94 symptomatic infants with perinatally acquired human immunodeficiency virus infection. Evidence for a bimodal expression of clinical and biological symptoms. Am. J. Dis. Child. 144: 12101215.
Brown, R. 1992. AIDS researchers focus on immune response. New Scientist 8 August, 16.
BuimoviciKlein, E., M. Lange, K.R. Ong, M.H. Grieco & L.Z. Cooper, 1988. Virus isolation and immune studies in a cohort of homosexual men. J. Med. Virol. 25: 371385.
Burke, D.S., 1992. Human HIV vaccine trials: Does antibodydependent enchancement pose a genuine risk? Perspectives Biol. Med. 35: 511531.
Burger, H. & B. Weiser, 1986. Transient antibody to human immunodeficiency virus. Ann. Intern. Med. 105: 464465.
Burger, H., B. Weiser, W.S. Robinson, J. Lifson, E. Engleman, et a/., 1985. Transient antibody to Lymphadenopathyassociated virus/human Tlymphotopic virus type 111 and Tlymphocyte abnormalities in the wife of a man who developed the aquired immonodeficiency syndrome. Ann. Int. Med. 103: 545547.
Calleq, M., 1990. Surviving AIDS. New York: Harper Collins.
Centers for Disease Control, 1993. HIV/AIDS Surveillance Report, June 1993.
Clerici, M., J.V. Giorg i, C.C. Chou, V.K. Gudeman, J.A. Zack, R Gupta, H.N. Ho, RG. Nishanian, J.A. Berzofsky & G.M. Shearer, 1992. Cellmediated immune response to human immunodeficiency virus (HIV) type in seronegative homosexual men with recent sexual exposure to HIVI. J. Infect. Dis. 165: 10121019.
Clerici, M. & G.M. Shearer, 1993. A THI -TH2 switch is a critical step in the etiology of HIV infection. Immunology Today 14: 107111.
Cohen, J., 1993. AIDS research: the mood is uncertain. Science 260: 12541255.
Cooper, M.H.. D. Maraninchi, J.A. Gastaut, P. Mannoni & Y. Carcassonne, l 993. HIV infection in autologous and allogeneic bone marrow transplant patients: a retrospective analysis of the Marseille bone marrow transplant population. J. Acquir. Immune. Defici. Syndr. 6: 277284.
Cuthbert, R.J.G., C.A. Ludlum, J. Tucker, er aL, 1990. Fve year
· prospective study of HIV infection in the Edinburgh haemophiliac cohon. Br. tried. J. 301: 956961.
Dalgleish. A.G., S. Wilson, M. Gompels, C. Ludlam, B. Gazzard, A.M. Coates & J. Habeshaw, 1992. Tcell receptor variable gene products and early HIVI infection. Lancet 339: 824828.
Daniel, V.,R. Weimer, K. Schimpf & G. Opeiz, 1989. Autoanubodies against CD4 and CD8positive T Lymphocytes in HlVinfected hemophilia patients. Vox Sang. 57: 172176.
Daniel, V., R. NVeimer, R. Zimmerman, A. HuthKuhne, et al., 1991. Improving CD4+ Lymphocyte counts in HlVinfected hemophilia patients: a favourable prognostic indicator? Immuno. Lett. 30: 2730.
Darby, S.C., R. Doll, B. Thakrar, C.R. Rizza & D.R. Cox. 1990. Time from infection with HIV to onset of AIDS in patients with haemophilia in the UK. Stat. Med. 9: 681689.
De Biasi, R., A. Rocino, E. Miraglia, L. Mstrulle & A.A. Quinno, 1991. The impact of very high purity factor VlII concentrate on the immune system of human immunodeficiency virusinfected hemophiliacs: a randomized prospective, twoyear comparison with an intermediate purity concentrate. Blood 78: 19191922.
Donohoe, R.hl. & A.A. Falek, 1988. Neuroimmunomodulation by opiates and other drugs of abuse: relationship to HIV infection and AIDS. In: T.R Bridge (ed.) Psychological, Neuropsychiatric, and Substance Abuse Aspects of AIDS. New York: Raven Press, 165172.
Downs, A.M., R.A. AncellePark, D. Costagliola, J.R Rigaut & J.B. Brunet.1991. Transfusionassociated AIDS cases in Europe: Estimation of the incubation period, distribution and prediction of future cases. J. Acquir. Immune. Defic. Syndr. 4: 805813.
Dudding, B.A., S.C. Wagner, J.A. Zeller, J.T. Gmelich, G.R. French & EH. Top, Jr., 1972. Fatal pneumonia associated with aden
ovirus type7 in three military trainees. New Engl. J. Med. 286: 1 289 1 292.
Duesberg, PH., 1987. Retroviruses as carcinogens and pathogens. Expectations and reality. Cancer Res. 47: 11991226.
Duesberg, RH., 1989. Human immunodeficiency virus and acquired immunodeficiency syndrome: Correlation but not causation. Proc. Natl. Acad. Sci. U.S A. 86: 755764.
Duesberg, P.H., 1990. AIDS: noninfectious deficiencies acquired by drug consumption and other risk factors. Res. Immunol. 141: 517.
Duesberg, P.H., 1992. The role of drugs in the ongin of AIDS. Biomed. & Pharmacother. 46: 315.
Drew, W.L., J. Mills, J. Levy, etaL, 1985.Cytomegalovirusinfection and abnormal Tlymphocyte subset ratios in homosexual men. Ann. Intem. Med. 103: 6163.
European Study Group on AIDS, 1989. Risk factors for male to female transmission of HIV. Br. Med. J. 298: 411415.
Eyster, M.E., M.H. Gail, I.O. Ballard, H. AlMondhiry & J.J. Goedert, 1987. Natural history of human immunodeficiency virus infections in hemophiliacs: effects on Tcell subsets, platelet counts and age. Ann. Intem. Med. 107: 16.
Farrugia, A., 1992. Purity of factor concentrates and immune function in hemophiliacs. Blood 78: 19191922.
Fauci, A.S., 1993. Multifactorial nature of human immunodeficiency virus disease: implications for therapy. Science 262: 101 11018.
Farzadegan, H., M.A. Polis, S.M. Wolinsky, C.R. Rinaldo Jr., J.J. Sninsky, S. Kwok, R.L. Griffiths. R.A. Kaslow. J.R Phair, B.F. Polk & A.A. Saah, 1988. Loss of human immunodeficiencyvirus type I (HIVI) antibodies with evidence of viral infection in asymptomatic homosexual men. Ann. Med. 108: 785790.
FemandezCruz, E., A.M. Femandez, C. Gunerrez, et al, 1988. Progressive cellular immune impairment leading to development of AIDS: Two year prospective study of HIV infection in drug addicts, Clin. Exp. Immunol. 72: 190195.
Fiala, M., L.A. Cone, C.M. Chang & E.S. Mocarski. 1986. Cytomegalovirus viremia increases with progressive immune deficiency in patients infected with HTLVIII. AIDS Res. 2: 175181.
FribourgBlanc, A., 1988. Deux observations d'annulation spontanee d'une seropositivite HIV. Med. Mall Infect. 4: 216218.
Fumento, M., 1990. The Myth of Heterosexual AIDS, New York, Harpers.
Garry, R.F., JJ. Kort, F. KochNolte & G. Koch, 1991. Tatcontams a sequence related to snake neurotoxins. AIDS. 5: 13811384.
Gibbons. J., J.M. Cory, I.K. Hewlen, J.S. Epstein & M.E. Eyster, 1990. Silent infections with human immunodeficiency virus type I are highly unlikely in multitransfused seronegative hemophiliacs. Blood 76: 19241926.
Giesecke, J., G.R ScaliaTomba, O. Berglund. E. Bemtorp, S. Schulman & L. Stigendal, 1988. Incidence of symptoms and AIDS in 146 Swedish haemophiliacs and blood transfusion recipients infected with human immunodeficiency virus. Br. Med. J. 297: 99102.
Goedert, J.J., C.M. Kessler, L.M. Aledort, et al., 1989. A prospective study of human immunodeficiency virus type I infection and the development of AIDS in subjects with hemophilia. New Engl. J. Med.321: 11411148.
Golding. H., EA. Robey, ET. Gates 111, W. Linder, et al., 1988. Indennfication of homologous regions in human immunodeficiency virus I gp41 and human MHC class 11 beta I domain. J. Exp. Med. 167, 914923.
Goldsmith, J.M., J. Deutsche, M. Tang & D. Green, 1991. CD4 cells in HIVI infected hemophiliacs: effect of Factor VIII concentrates. Thromb. Haemost. 66: 415419
Gonlieb, M.S., R.A. Zackin, M. Fiala, D.H. Henry, A.J. Marcel., K.L. Combs, J. Vieira, et al, 1991. Response to treatment with the leukocytederived immunomodulator IMREGI in immunocompromised patients with AlDSrelated complex. Ann. Intem. Med. 115:8491.
Gomperts, E.D., R. De Biasi & R. De Vreker, 1992. The impact of cloning factor concentrates on the immune system in individuals with hemophilia. Transfus. Med. Rev. 6: 4454.
Gougeon, M.L. & L. Montagnier, 1993. Apoptosis and AIDS. Science260: 12961271.
Groenbladh, L. & L. Gunne, 1989. Methadoneassisted rehabilitation of Swedish heroin addicts. Drug Alcohol Depend. 24: 3137.
Gurtler, L., J. Eberle & E Deinhardt, 1989. Seroreversion in human immunodeficiencyvirus (HIV) infection. Ann. Intem. Med. 110: 946948.
Hadden, J.W., 1991. Immunotherapy of human immunodeficiency virus infection. Trends Pharmcol. Sci. 12: 1071 1 1.
Halstead, S.B., 1988. Pathogenesis of dengue: Challenges to molecular biology. Science 29: 476481.
Hamilton, J.R., J.C. Overall & L.A. Glasgow. 1976. Synergistic effect on mortality in mice with munne cytomegalovirus and Pseudomonas arrugionsa, Staphylococcus aureus, or Candida albicans infections. Infect. Immunol.14: 928989.
Hardy, A.M., J.R. Allen, W.M. Morgan & J.W. Curran, 1985. The incidence rate of acquired immune deficiency syndrome in selected populations. JAMA 253: 215218.
Hassen, J., G.E Gjerset, J.W. Mosley, M.A. Fletcher, et al., 1993. Effect on Lymphocyte subsets of cloning factor therapy in human immunodeficiency virus I negative congenital cloning disorders. Blood 82: 13511357.
Haverkos, H.W., 1988. Kaposi's sarcoma and nitrite inhalants. In: T.R Bndge (ed.) Psychological, Neuropsychiatric, and Substance Abuse Aspeas of AIDS. New York: Raven Rress, 165 172.
Hay, C.R.. R McEvoy & M. DugganKeen, 1990. Inhibitions of Lymphocyte IL2receptor expression by factor VIII concentrate: a possible cause of immune suppression in haemophiliacs. Br. J. Haematol. 75: 278281.
Hendricks, J.C., G.E Medley, G.J. Van Griensven, R.A. Coutinho, et al, 1993. The treatmentfree incubation period of A'DS in a cohort of homosexual men. AIDS 7: 231239.
Henrard, D.R.Y. Laurian, W.E Mehaffey & J.R Allain, 1993. Lack of detection of human immunodeficiency virus type I DNA by
polymerase chain reaction in the plasma and Lymphocytes of seronegative exposed hemophiliacs. Transfusion 33: 405408.
Higgins, J.A. & A.H. Goodall, 1991. Immune pctivation in HIVseronegative hemophiliacs: Is this related to infection with hep atitis C virus? Thromb. Haemost.65: 1163 (abstr. 1682).
Hilgartner, M.W., J.D. Buckley, E.A. Operskalsi, M.C. Pike & J.W. Mosely, 1993. Purity of factor VIII concentrates and serial CD4 counts. Lancet 341: 1373 1374.
Hoff, C. & R.D.A. Peterson, 1989. Does exposure to HLA alloantigens trigger immunoregulatory mechanisms operative in both pregnancy and AIDS? Life Sci. 45: iiiix.
Hoff, C., W.C. James, R.B. Hester, R Nolan & R.D.A. Peterson, 1991. Signs of cellular immunosuppression correlate w ith HLADR phenotypes in healthy HlVnegative homosexuals: preliminary findings. Human Biology 63: 129135.
Holman, R.C., RH. Rodes, T.L. Chorba & B.L. Evan, 1992. Survival of hemophiliac males with acquired immunodeficiency syndrome with and without risk factors for AIDS other than hemophilia. Am. J. Hematol. 39: 275282.
Hoover, D.R., AJ. Saah, H. Bacellar, et al, 1993. Clinical manifestation of AIDS in the era of pneumocystis prophylaxis. New Engl. J. Med.329: 19221926.
Horsburgh, C.R., Jr., C.Y. Ou, J. Jason, et, al, 1990. Concordance of polymerase chain reaction with human immunodeficiency virus antibody detection. J. Infect. Dis. 162: 542545.
Huang, S.W. & R. Hong, 1973. Lymphopenia and multiple viral infections. IAMA 225: 11201121.
Huang, S.W., D.B. Lados, D.B. Nelson, K. Reb & R. Hong, 1973. Antibodyassociated lymphotoxin in acute infection. J. Clin. Invest. 52: 10331040.
Hughes, M D., D.S. Stein. H.M. Gundacker, ET. Valentine, J.R Phair & PA. Volberding, 1994. Withinsubject variation in CD4 lymphocyte count in asymptomatic human immunodeficiency virus infection: Implications for patient monitoring. J. Infect Dis. 169: 2836.
Imagawa, D. & R. Detels, 1991. HIVI in seronegativehomosexual men. NEJM 325:12501251.
Jaffe, H.W., RM. Feorino, W.W. Darrow, es aL, 1985. Persistent infection with human Tlymphotropic virus type III/lymphadenopathyassociated virus in apparently healthy homosexual men. Ann. Intern. Med. 102:627.
Judson, EN., 1990. The relationship of HIV infections to infections with pathogenic Neisseria in homosexual men. Med. Clin. North. Am. 74: 13531366.
Kallenius, G., S.E. Hoffner & S.B. Svenson, 1989. Does vaccination with bacille CalmetteGuerin protect against AIDS? Rev. Infect. Dis. 11:349351.
Keay, S., C.O Tacket, J.R. Murphy & B.S. Handv.erger, 1992. AntiCD4 antiidiotype antibodies in volunteers immunized with rgpl60 or HIVI or infected with HIVI. AIDS Res. Human Retroviruses8: 10911098.
Kion, T.A. & G.W. Hoffmann, 1991. AntiHIV and antiantiMHC antibodies in alloimmune and autoimmune mice. Science 253: 11381140.
Klenk, H.D. & R. Rott, 1988. The molecular biology of influenza virus pathogenicity. Adv. Virus Res. 34: 247281.
Kliks, S.C., A. Nisalak, W.E. Brandt, et aL, 1989. Antibodydependentenhancementofdenguevirusgrowthinhumanmono cytes as a risk factor for dengue hemorrhagic fever. Am. J. Trop. Med. Hyg. 40: 444451.
KnuverHopf, J., H. Heinze, B. Lambrecht, H. Mohr, J. Beyer & H. Schmitt, 1993. Blood donations indeterminate in HIVI Western blot analysed by IgM immunoblot and polymerase chain reaction. Vox Sang. 64: 8993.
KopecSchrader, E., B. Tindall, J. Learmont, B. Wylie & J.M. Kaldor, 1993. Developmentof AIDS in people with transfusionacquired HIV infection. AIDS 7: 10091013.
Krasinski. K., W. Borkowsky & R.S. Holzman, 1989. Prognosis of human immunodeficiency virus infection in children and adolescents. Pediatr. Infect. Dis. J. 8: 216220.
Lafeuillade, A. & R. Quilichine, 1992. HIV cofactors in the course of AIDS. Presse Med. 21: 14261430.
Lang, Ph., R Niaudet, and the Groupe Cooperatif de Transplantation de l'lle de France, 1991. Update and outcome of renal transplant patients with human immunodeficiency virus. Transplantation Proc.23: 13521353.
Laurence, J., 1993. Tcell subsets in health, infectious disease, and idiopathic CD4+ T Lymphocytopenia. Ann. Intem. Med. 119: 5562.
Levy, J., 1988. The transmission of AIDS: the case of the infected cell. JAMA 259: 30373038.
Littlefield. J.W., 1992. Possible supplemental mechanisms in the pathogenesis of AIDS. Clin. Immunol. Immunopathol. 65: 85q7
Lo, S.H., S. Tsai, J.R. Benish, et aL, 1991 . Enhancement of HIVI cytocidal effects in CD4+ Lymphocytes by the AlDSassociated mycoplasma. Science 251: 10741076.
Lorenzo, 11., R. Molina & J.A. Aznar, 1993. Evolution of CD4 and CD8 Lymphocyte subsets in HIV seronegative and seropositive hemophiliacs. Sangre Barc. 38: 511.
Ludlum, C.A., J. Tucker, C.M. Steel, et aL, 1985. Human Tlymphotropic virus type 111 (HTLV111) infection in seronegative hemophiliacs affertransfusions offactorVIII. Lancet ii: 233236.
Lui, K.J., D.N. Lawrence, W.M. Morgan, T.A. Peterman, H.W. Haverkos & D.J. Bregman, 1986. A model based approach for estimating the mean incubation period of transfusionassociated acquired immunodeficiency syndrome. Proc. Natl. Acad. Sci. U.S.A. 83: 301305.
Lusso, R. R. Ensoli, RD. Markham, et uL, 1989. Productive dual infection of human CD4+ T Lymphocytes by HIV I and HHV6. Nature (Lond.), 337: 370373.
Lusso, P., E Lori & R.C. Gallo, 1990. CD4independent infection by human immunodeficiency virus type I aher phenotypic mixing with human Tcell leukemia viruses. J. Virol. 64: 63416344.
Macon, W.R., S.C. Lo, B.J. Poisz, D.C. Monefiori, et al., 1993. Acquired immunodeficiencysyndromelike illness associated with systemic Mycoplasma fermentans infection in a human immunodeficiency virusnegative homosexual man. Human Pathol 24:
Madhok, R., J.A. Gracie, J. Smith, G.D. Lowe & C.D. Forbes, 1990. Capacity to produce interleukin 2 is impaired in haemophilia in the absence and presence of HIV I infection. Br. J. Haematol.76: 7074.
Madhok, R., J. Smith, A. Jenkins & G.D.O. Lowe, 1991. Tcell sensitization to factor Vlll in haemophilia A? Br. J. Haematol. 79: 235238.
Mannucci, RM., A. Gringeri, R. De Biasi, E Baudo, M. Morfini & N. Ciavarella, 1992. Immune status of asymptomatic HlVinfected haemophiliacs: randomized, prospective, twoyear comparison of treatment with highpunty or an intermediatepunty factor Vlll (FVIII). Thromb. Haemost.67: 31~313.
Mares, M., M.T. Sartori & A. Girolami, 1992. Prevalence of bleeding as cause of death in HlVseropositive patients with congenital cloning disorders. Vox Sang. 63: 7778.
Mathe, G., 1992. Is the AIDS virus responsible for the disease? Biomed. Pharmacother. 46: 1 2.
McClean, A.R. & M.A. Nowak, 1992. Models of interactions between HIV and other pathogens. J. Theor. Biol. 155: 6986.
McDougal, J.S., M. Hubbard, J.K.A. Nicholson, et al., 1985. Immune complexes in the acquired immunodeficiency syndrome (AIDS): relanonship to disease manifestation, nsk group, and immunologic defect. J. Clin. Immunol.5: 130.
Medawar, RB., 1967. The art of the soluble. London: Methuen.
Medley, G.E, R.M. Anderson, D.R. Cox & L. Billard, 1987. Incubation period of AIDS in patients infected via blood transfusion. Nature 328: 719721.
Mills, L.B., 1986. Stimulation of Tcellular immunity by cutaneous application of dinitrochlorobenzene. J. Am. Acad. Dermatol. 14: 10891090.
Minkoff, H., D. Nanda, R. Menez & S. Flkrig, 1987. Pregnancies resulting in infants with acquired immunodeficiency syndrome or AlDSrelated complex: followup of mothers, children, and subsequently born siblings. Obstet. Gynecol. 69: 288291.
Montagnier, L., D. Berriman, D. Guetard, A. Blanchard, et al., 1990. Inhibition de l'infectiosite de souches prototype du VIH par des anticorps diriges contre une sequence peptideque de mycoplasme. Comptes rendus Acad. Sci. Paris 311: 425430.
Montaner, J.S., L M. Lawson, A. Gervais, et al., 1991. Aerosol pentamidine for secondary prophylaxis of AlDSrelated PneumfJCystis cariniipneumonia. A randomized, placebocontrolled study. Ann. Intern. Med. 114: 948953.
Moreni, M.L., 1992. Specific behavioural factors among intravenous drug users have been shown to influence HIV seroconversion . . . J. InterAm. Med. Health. Assoc. 1: 18.
Morrow, W.J.W,, D.A. Isenberg, R.E. Sobol, R.B. Stricker & T. KeiberEmmons, 1991. AIDS virus infection and autoimmunity: A perspective on the clinical, immunological, and molecular origins of the autoallergic pathologies associated with HIV disease Clin. Immunol. Immunopathol. 58, 163180.
Msellati, R. M. Dupon, R Morlat, D. Lacoste, er al., 1990. A cohort study of 89 HIV I infected adult patients contaminated by blood products: Bordeaux 19811989. AIDS 4: 1 1051 109.
Munoz, A., V. Carey, A.J. Saah, et al., 1988. Predictors of decline in CD4 lymphocytes in a cohort of homosexual men infected with human immunodeficiency virus. 1. Acquir. Immune. Defic. Syn. 1:396.
National Research Council. 1990. AIDS: The Second Decade National Academy of Sciences, Washington, D.C.
Naz, R.K., M. Ellauri, T.M. Phillips & J. Hall, 1990. Antisperm antibodies in human immunodeficiency virus infection: Effects on fertilization and embryonic development. Biol. Reprod. 42: 859868.
Odell, T.W. & J.A. Green, 1990. Prolonged zidovudine therapy: confounded by Pneumocysitis carinii prophylaxis? JAMA 263: 1 635.
Oehen, S., H. Henganner & R.M. Zinkemagel, 1991. Vaccination for disease. Science 251: 195198.
Ozturk, G.E., RE Kohler, C.R. Horsburgh & C.H. Kirkpatrick, 1987. The significance of antilymphocyte antibodies in patients with acquired immune deficiency syndrome (AIDS) and their sexual pawners. J. Clin. Immunol. 7: 130133.
Padian, N.S., S.C. Shiboski & N.R Jewell, 1990. The effect of number of exposures on the risk of heterosexual HIV transmission. J. Infect. Dis. 161: 883887.
Palestine, A.G., M.A. Polis, M.D. De Smet, et al., 1991. A randomized, controlled trial of foscarnet in the treatment of cytomegalovirus retinitis in patients with AIDS. Ann. Intem. Med. 115: 665671.
PapadopulosEleopulos, E., 1988. Reappraisal of AIDS. Is the oxidation induced by the risk factors the primary cause? Med. Hypotheses25: 151162.
Perrin, L.H., R. Zubler, B. Hirschel, J.L. Manin, D. Salomon, J.H. Sauret & O. Fryc, 1988. Negativation d'une serologic positive pour le virus dtimmunodefience humaine (VIH). A propos de deux observations. Schweiz. Med. Wochenschn. 118/45: 16411644.
Peterman, T.A., R.L. Stoneburner, J.R. Allen, H.W. Jaffe & J.W. Curran. 1988. Risk of human immunodeficiency virus transmission from heterosexual adults with transfusionassocinted infections. JAMA 259: 5558.
Pifer, L.W., Y.F. Wang, T.M. Chiang, R. Ahokas, D.R. Woods & R.E. Joyner, 1987. Borderline immunodeficiency in male homosexuals: Is lifestyle contributory? South. Med. J. 80: 687697.
Popper, K., 1962. Conjectures and refutations. The growth of scientific knowledge. New York: Basic Books.
Ponerfield, J.S., 1986. Antibodydependent enhancement of viral activity. Adv. Virus. Res. 31: 335353.
Rahman, M.A., L.A. Kingsley, M.K. Berinig, et aL, 1989. Reactivation of EpsteinBarr virus during early infection with human immunodeficiency virus. J. Clin. Microbiol. 29: 12151220.
Ribot, S. & H.H. Eslami, 1992. HIV infection in kidney transplant recipients. N. J. Med. 89: 597599.
Rinaldo. C.R. Jr., L.A. Kingsley, D.W. Lyter, et al., 1986. Association of HTLVII I with EpsteinBarr virus infection and abnormalities of T Lymphocytes in homosexual men. J. Infect. Dis. 154: 556561.
Rinaldo, C.R., Jr., L.A. Kingsley, M. Ho, J.A. Armstrong & S.Y.J. Zhou, 1992. Enhanced shedding of cytomegalovirus in semen of human immunodeficiency virusseropositive homosexual men. J. Clin. Microbiol. 30: 11481155.
Riner, M., 1994. AIDS raises death rate for hemophilia. Associated Press release based on CDC press conference, 3 Feb. 1994.
Roithmann, S., J.M. Tourani, J.A. Gastaut, R Brice, er al., 1993. Hodgkin's disease associated with HIV infection: clinical charactenstics and development. Bull. Cancer. Paris. 79: 873882.
RootBemstein, R.S., 1989. Discovering, Inventing and solving problems at the frontiers of knowledge. Cambridge, MA: Harvard University Press.
RootBemstein, R.S., 1990a. Do we know the cause(s) of AIDS? Persp. Biol. Med. 33: 480500.
RootBemstein, R.S., 1990b. Multipleantigenmediated autoimmunity (MAMA) in AIDS: a possible model for postinfectious autoimmune complications. Res. Immunol.141: 321339.
RootBemstein, R.S., 1990c. NonHIV immunosuppressive agents in AIDS: a multifactorial, synergistic theory of AIDS aetiology. Res. Immunol. 141: 815838.
RootBemstein, R.S., 1991. Self, nonself, and the paradoxes of autoimmuniry. In: All. Tauber, ed. Organism and the Development of Self. Kluwer Academic, pp.159209.
RootBemstein, R.S., 1992a.HIV and immunosuppressivecofactors in AIDS. EOS. Rev. Immunol. Immunopharm.12: 256262.
RootBemstein, R.S., 1992b. AIDS is more than HIV. Genetic Engineenng News 12 no. 13: 46; 12 no. 14: 45.
RootBemstein, R.S., 1993. Rethinking AIDS. The Tragic Cost of Premature Concensus. New York: Free Press.
RootBemstein, R.S., 1994. Preliminary evidence of idiotypeantiidiotype immune complexes crossreactive w ith Lymphocyte antigens resulting from pairs of infectious agents associated with AIDS and lupus. Med. Hypoth. In press.
RootBemstein, R.S. & S.H. Hobbs. 1991. Homologies between mycoplasmaadhesion pepetide, CD4, and class 11 MHC proteins: a possible mechanism for HlVmycoplasma synergism in AIDS. Res. Immunol.142:519523.
RootBemstein, R.S. & S.H. Hobbs, 1993. Does HIV 'piggyback'on CD4like surface proteins of sperm, viruses, and bactena? Implications for cotransmission, cellular tropism and the induction of autoimmunity in AIDS. J. Theor. Biol. 160: 249264.
RootBemstein, R.S. & S.H. DeWitt, 1994. CD4 similarity to proteins of infectious agents in AIDS, and autoimmunity. Med. Hypoth. In press.
Rosendaal, EC., C. Smit & E. Briet, 199 B Hemophilia treatment in historical perspective: a review of medical and social developments. Ann. Hematol. 62: 525.
Roy, M.J., JJ. Damato & D.S. Burke, 1993. Absence of true seroreversion of HIVI antibody in seroreactive individuals. JAMA 269: 28762879.
Rubenstein, A., M. Sicklick, A. Gupta, L. Bemstein, N. Klein, et al., 1983. Acquired immunodeficiency with reversed T4/T8 ratios in infants bom to promiscuous and drugaddicted mothers. IAMA 249: 23502356.
Rubin, H., 1988. Etiology of AIDS. Science 214: 13891390.
Sabin.A., 1988.Effectiveness of AIDS vaccines. Science 251: 1161.
Sabin,C.,A. Phillips, J. Elford. R Griffiths, G. Janossay & C. Lee, 1993. Prcrgression of HIV disease in a haemophiliac cohort followed for 12 years. Br. J. Haemoatol.83: 330335.
Salk, I.P.A. Bretscher, RL. Salk, M. Clerici & G.M. Shearer, 1993. A strategy for prophylactic vaccination against HIV. Science 260: 12701272.
Schinaia, N., A. Ghirardini, E Chiaroni, A. Gringeri, PM. Mannucci, et al, 1991. Progression of AIDS among Italian HIVseropositive haemophiliacs. AIDS 5/4: 385391.
Schulman. S., 1991. Effects of factor VIII concentrates on the immune system of hemophiliac patients. Ann. Hematol.63; 145151.
Schwarz. A., G. Offerman, E Keller, 1. Bennhold, J. L'ageStehr, RH. Krause & M.J. Mihatsch, 1993. The effect of cyclosporine on the progression of human immunodeficiency virus type I infection transmitted by transplantation data on four cases and review of the literature. Transplantation 55: 95103.
Shearer, G.M. & R.B. Levy. 1984. Noninfectious cofactors in susceptibility to AIDS: possible contributions of semen, HLA alloantigens, and lack of natural resistance. Ann. N. Y. Acad. Sci. 437: 4957.
Shope, E., 1931. Swine influenza, 111. Filtration experiments and etiology. J. Exp. Med.54: 373385.
Sonnabend. I.A., 1989. AIDS: An explanation for its occurrence in homosexual men. In: AIDS and Opportunistic Infections of Homosexual Men, R Ma and D. Armstrong, eds. Bunerworth Publishers, Stoneham. MA, pp. 450460.
Sonnabend. J.A. & S. Saadoun, 1984. The acquired immunodeficiency syndrome: A discussion of etiologic hypothesis. AIDS Res. 1: 107120.
Sonnabend, J.A., S.S. Witkin & D.T. Portillo, 1984. A multifactorial model for the development of AIDS in homosexual men. Ann. N. Y. Acad. Sciences 437: 177182.
Sorice, E. A. Ghirardini, P. Martino, D. Pasqualeni, et aL, 1989. Spectrum of HIV infection and AIDS in a cohort of Italian hemophiliacs. Haemotologica74: 149154.
StehrGreen, J.K., J.M. Jason, B.L. Evan, R.C. Holman, et aL, 1989. Geographic variability of hemophiliaassociated AIDS in the United States: Effect of population characteristics. Am. J. Hematol.32: 178183.
Ston, E.J., 1991. Anticell antibody in macaques. Nature 353: 393395.
Stricker, R.B.. T.M. McHugh, DJ. Moody, W.J.M. Morrow, D.R Stites, M.A. Shuman & J.A. Levy, 1987a. An AlDSrelated cytotoxic antoantibody reacts with a specific antigen on stimulated CD4+ cells. Nature 327: 710713.
Stricker, R.B.. T.M. McHugh, RA. Marx, W. J.M. Morrow, J.A. Levy, M.A. Shuman, D.R Stites & RD. Nyeman,1987b. Prevalence of an AlDSrelated autoantibody against CD4+ Tcells in humans and monkeys. Blood 70: 127A.
Stricker, R.B., B.E Elswood & D.l. Abrams, 1991. Dendritic cells and dinitrochlorobenzene (DNCB): a new treatment approach to AIDS. Immunol.Len.29: 191196.
Stricker, R.B., B.E Elswood, D.l. Abrams, et aL, 1993. Pilot study of topicaJ dinitrochlorobenzene (DNCB) in human immunodeficiency virus infection. Immunol. Len. 36: 16.
Sullivan, J.L.. EE. Brewster, D.B. Brettler, ctaL, 1986. Hemophiliac immunodeficiency: Influence of exposure to factor Vlil concentrate, LAVHTLV111, and herpes viruses. J. Pediatr. 108: 504510.
Sumaya, C.V., R.N. Boswell, Y. Ench, et aL, 1985. Enhanced serological and virological findings of EpsteinBarr virus in patients with AIDS and AIDS related complex. J. Infect Dis. 154: 864.
Susal, C., M. Krdpelin, V. Daniel & G. Opeiz, 1993. Molecular mimicry between HIVI and antigen receptor molecules: a clue to the pathogenesis of AIDS. Vox Sang. 65: 1017.
Tanenbaum, S.A., R.E Garry, H. LuoZhang, S.S. Alexander & C.A. Leissinger, 1993. Humoral and cellular responses against HIV in multitransfused HIV seronegative hemophiliacs. Absts Ist National Conf. Human Retroviruses. Abst. #86, p. 71.
Tanenbaum, S.A., C.A. Leissinger & R.E Garry. 1993. Absence of seroconversion of HIVI antibody in seroreactive individuals. JAMA 270:2178.
Taylor, J.M., J.M. Duo & R. Detels, 1991.1s the incubation period of AIDS lengthening? J. Acquir. Immune. Defic. Syndr. 4 6975.
Tzakis, A.G., M.H. Cooper, J.S. Dummer, M. Ragni, J.W. Ward & T.E. Starzl, 1990. Transplantation in HIV+ patients. Transplantation 49: 354358.
Umovitzj H.B.. M. Clerici, G.M. Shearer, T.D. Gonfned, D.J. Robinson, L.l. Lutwick, L. Montagnier & D.V. Landers, 1993. HIVI antibody serum negativity with urine positivity. Lancet 342: 14581459.
Vamvakas, E. & H.S. Kaplan, 1993. Early transfusion and length of survival in acquired immune deficiency syndrome: experience with a population receiving medical care at a public hospital. Transfusion 33: 111118.
Vandenbroucke, J.R & V.RA.M. Pardoel, 1989. An autopsy on epidemiologic methods: the case of 'poppers' in the early epidemic of the acquired immunodeficiency syndrome (AIDS). Am. J. Epidem. 129:455457.
Van Griensven, G.J., E.M. de Vroom, J. Goodsmit & R.A. Coutinho, 1989. Changes in sexual behaviour and the fall in incidence of HIV infection among homosexual men. Br. Med. J.298: 2 18221.
Vega, M.A., R. Guigo & T.F. Smith, 1990. Autoimmune response in AIDS. Nature 345:26.
Wang, M.C. & L.C. See, 1992. Nestimation from retrospectively
as certained events with application to AIDS. Biometrics48: 129141.
Wang, R.Y., J.\V. Shih, T. Grandineni, RE Pierce, M.M. Hayes, D.J. Wear, H.J. Alter & S.C. Lo, 1992. High frequency of antibodies to Mycoplasma penetrans in HlVinfected patients. Lancet 340: 13121316.
Ward, J.W., T.J. Bush, H.A. Perkins, et aL, 1989. The natural history of transfusionassociated infection with human immunodeficiency virus. New Engl. J. Med. 321: 947952.
Watson, H.G. & C.A. Ludlum, 1992. Immunological abnormalities in haemophiliacs. Blood Rev. 6: 2633.
Weber, R., W. Ledergerber, M. Opravil, W. Siegenthaler & R. Luthy, 1990. Progression of HIV infection in misusers of injected drugs who stop injecting or follow a programme of maintenance treatment with methadone. Br. Med. J. 301: 1361 1365.
Webster, A., C.A. Lee, D.G. Cook, J.E. Grundy, et al., 1989. Cytomegalovirus infection and pregression towards AIDS in haemophiliacs with human immunodeficiency virus infection. Lancet ii: 6366.
Weiss, R.A., 1993. How does HIV cause AIDS? Science 260: 12731279.
Weyer, J. & H.J. Eggers, 1990. On the structure of the epidemic spread of AIDS: the influence of an infectious coagent. Int. J. Med. Microbiol.273: 5267.
Winkelstein, \V. Jr, J.A. Wiley, N.S. Padian, M. Samuel, S. Shiboski, M.S. Ascher & J.A. Levy.1988. The San Francisco Men's Health Study: Continued decline in HIV seroconversion rates among homosexual/bisexual men. Am. J. Public Health 78: 14721474.
Witebsky, E., N.R. Rose, K. Terplan, J.R. Pain & R.W. Egan, 1957. Chronic thyroiditis and autoimmunization. JAMA 164: 1439
Zarling, I.M., J.A. Ledbetter, J. Sias, P. Fultz, J. Eichberg, G. Gjerset & RA. Moran, 1990. HlVinfeeted humans but not chimpanzees, have circulating cytotoxie T Lymphoeytes that Lyse uninfected CD4+ cells J. Immunol. 144. 29922998.
Ziegler, J.L. & D.P. Stites, 1986. Hypothesis: AIDS is an autoimmune disease directed at the immune system and triggered by a Lymphotropie retrovirus. Clin. Immunol. Immunopathol. 41: 305.
Zuck, T.F., 1988. Silent sequences and the safety of blood transfusion Ann Inter. Med. 108 895896.